高危神經母細胞瘤(NB)常涉及MYCN擴增和ALK突變。目前,高危NB存在重大的臨床挑戰,需要更多的治療選擇。
瑞典和比利時的科學家合作在Nature Communications (中科院JCR一區,影響因子:14.919)上發表了題為“ATR inhibition enables complete tumour regression in ALK-driven NB mouse models”的論文。通過磷酸化蛋白組學結果發現ALK驅動的NB細胞中存在著ATR的激活,抑制ATR可顯著阻礙腫瘤細胞生長和增殖,甚至可以使ALK驅動的神經母細胞瘤完全消退。最后通過RNA測序、蛋白質組學和磷酸化蛋白組學,表征了NB細胞和腫瘤對ATR抑制的應答,將DNA損傷反應的關鍵成分確定為NB 細胞中的 ATR 靶點。本研究為高危神經母細胞瘤的治療提供了一個潛在的機會。
研究思路
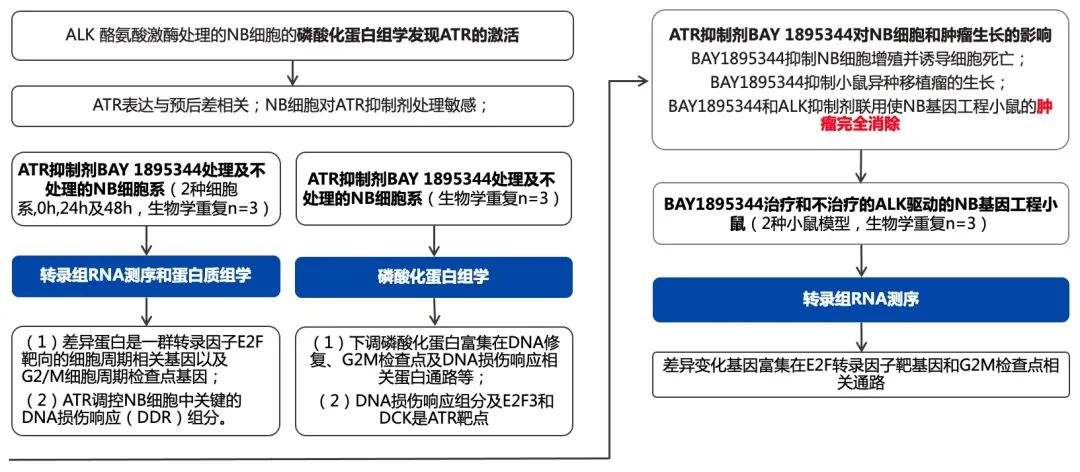
1. 磷酸化蛋白質組學:ATR是ALK信號的靶點
作者此前用ALK酪氨酸激酶抑制劑(TKI)處理ALK驅動的NB細胞,并通過磷酸化蛋白組學檢測發現了ALK抑制后會導致Sad1和SUN2蛋白N端的絲氨酸和蘇氨酸磷酸化的顯著變化。此外,ATR絲氨酸435、436和437的磷酸化也下調。而抗ATR/ATM底物抗體的富集產物中發現了SUN2磷酸化,這增加了NB細胞中ALK活性調節ATR/ATM活性的可能性。進一步,在NB細胞中,抗pALK/ATM底物motif的抗體富集后能檢出SUN2,而不管是用ALK酪氨酸激酶抑制劑lorlatinib,還是ATR抑制劑BAY 1895344處理NB細胞,抗體IP產物中檢出的SUN2都減少。這些結果意味著,ALK驅動的NB細胞中存在著ATR的激活。
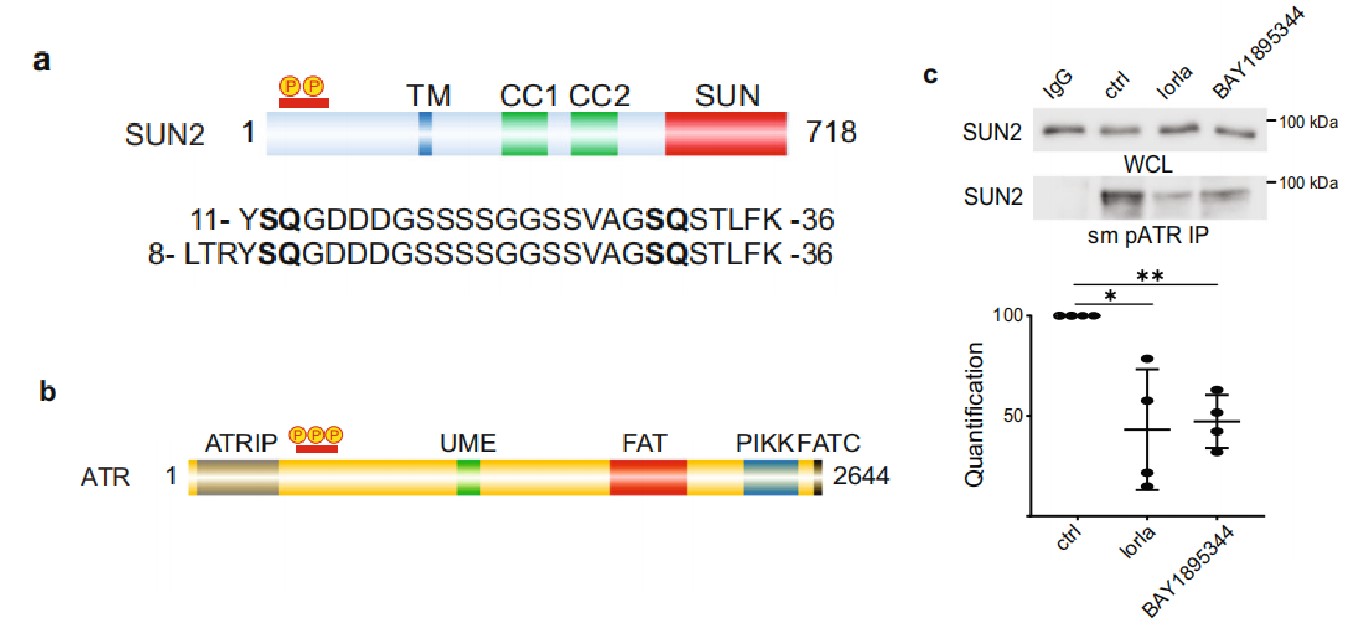
2. ALK驅動的NB細胞系對ATR抑制敏感
作者分析了一個已發表的NB數據,發現高表達的ATR與預后差具有相關性。作者繼續在9個不同基因背景的NB細胞系中檢測ATR/pATR和相關的ATM/pATM及下游信號組分FOXM1/pFOXM1 和CHK1/pCHK。所有NB細胞系中均檢測到了pATR、 pATM、pFOXM1和 pCHK1。這意味著這些細胞系存在著基礎的DNA損傷響應(DDR)。隨后,作者用ATR抑制劑BAY 1895344處理兩個由ALK驅動的NB細胞系,這兩個細胞系均對ATR抑制劑表現出高敏感性。
3. ATR抑制劑阻礙NB細胞生長并不受細胞ALK和MYCN狀態的影響
前面的研究表明,pATR在NB細胞系中廣泛表達,包括非ALK驅動的NB細胞。作者進一步在非ALK驅動的NB細胞中檢測ATR抑制劑對細胞生長的影響。結果顯示,所有的非ALK驅動的NB細胞也對ATR抑制劑BAY1895344敏感。同時,作者還發現,在ATR抑制劑造成了DNA損傷響應相關蛋白和凋亡的上調,該結果在siRNA介導的ATR敲低 NB細胞中得到進一步證實。
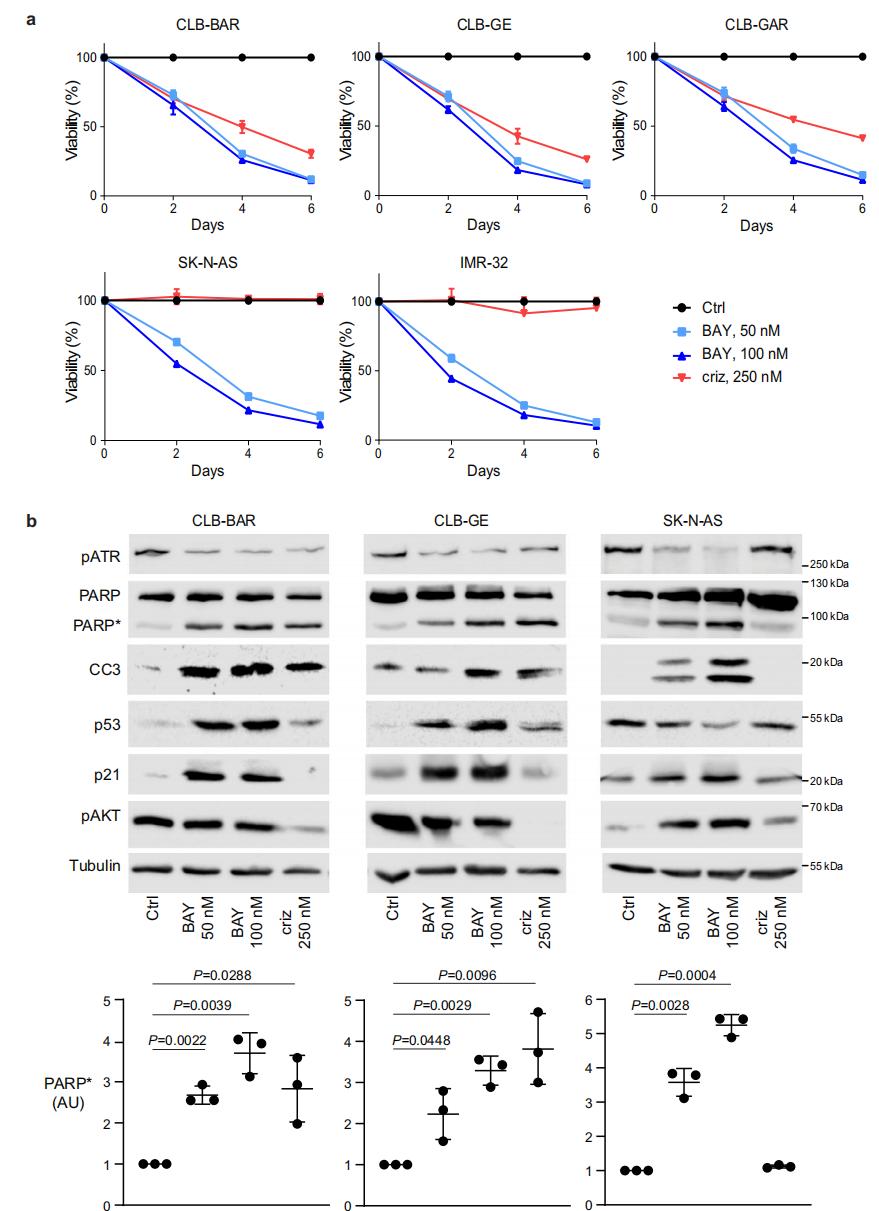
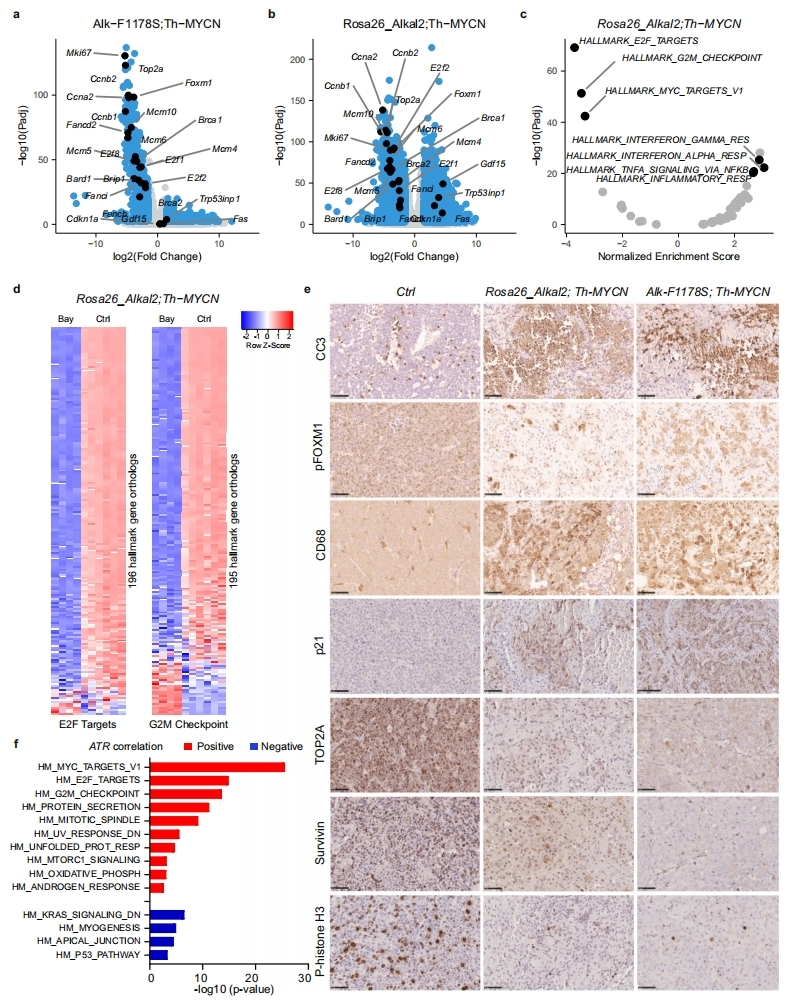
4. ATR抑制的NB細胞的轉錄組和蛋白質組變化
為了研究抑制ATR后的下游基因表達響應,作者分別對ATR抑制劑BAY1895344處理0h、24h和48h的NB細胞(CLB-BAR和CLB-GE細胞系,均受ALK驅動)進行轉錄組和蛋白質組學檢測。轉錄組結果顯示,CLB-GE細胞在處理48h后顯示出最多的基因表達變化(370個上調,198個下調)。這些下調基因幾乎都在48h才顯示出變化,它們主要是一群轉錄因子E2F靶向的細胞周期相關基因以及G2/M細胞周期檢查點基因。上調基因包含了一群富集在p53通路的基因。蛋白質組學呈現了類似的結果:基于GSEA的轉錄因子預測確認了E2F轉錄因子的關鍵作用,同時發現了其他轉錄因子,如參與DNA損傷響應(DDR)的RAD51和BRCA1/2。
作者監測了細胞周期過程中ATR的下游靶點pFOXM1(T600)和pChk1(S345)的水平。
結果表明,胸腺嘧啶阻滯解除后,在BAY1895344抑制的CLB-BAR細胞中,pFOXM1表達增強。在BAY1895344抑制的CLB-GE細胞中,觀察到pFOXM1和pCHK1的類似變化。同時還發現,與ATR抑制相比,ALK抑制劑對pFOXM1沒有影響。因此,在NB細胞中,ATR對S/G2檢查點的調控是保守的。
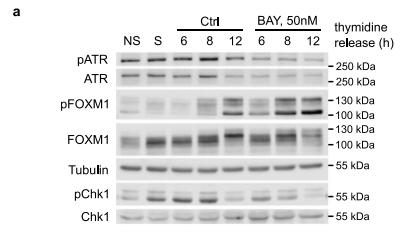
為了進一步研究ATR抑制劑BAY1895344對NB細胞的影響,作者使用抑制劑對從胸腺嘧啶阻滯解放出來的CLB-BAR細胞進行處理,并檢測處理6h后的磷酸化蛋白質組學變化。共鑒定到4087個蛋白上的14528個磷酸化位點。ATR自磷酸化位點T1989以及另外的S435都發生了去磷酸化。其他磷酸化下調的ST/Q位點包括AEBP2 S167、 UTP14A S445、DCK S74 和CASC3 S10 。非ST/Q位點包括ATR底物 FANCD2 T716、E2F3 S172/166、 SLBP S110 和TOPBP1 S888。特別地,下調磷酸化位點中還發現了DNA損傷響應相關蛋白,如ATRX、FANCI、EXO1和WRN。下調磷酸化位點富集在DNA修復及相關通路如G2M檢查點和范康尼貧血通路。作者的磷酸化蛋白組同時發現,大量下調的磷酸化位點并不包含ATR底物的S/TQ motif,這意味著,蛋白磷酸化的變化并不是一種直接的調控,而是二級調控結果。總體,磷酸化蛋白質組學發現了DNA損傷響應組分及E2F3和DCK是ATR作用的靶點。
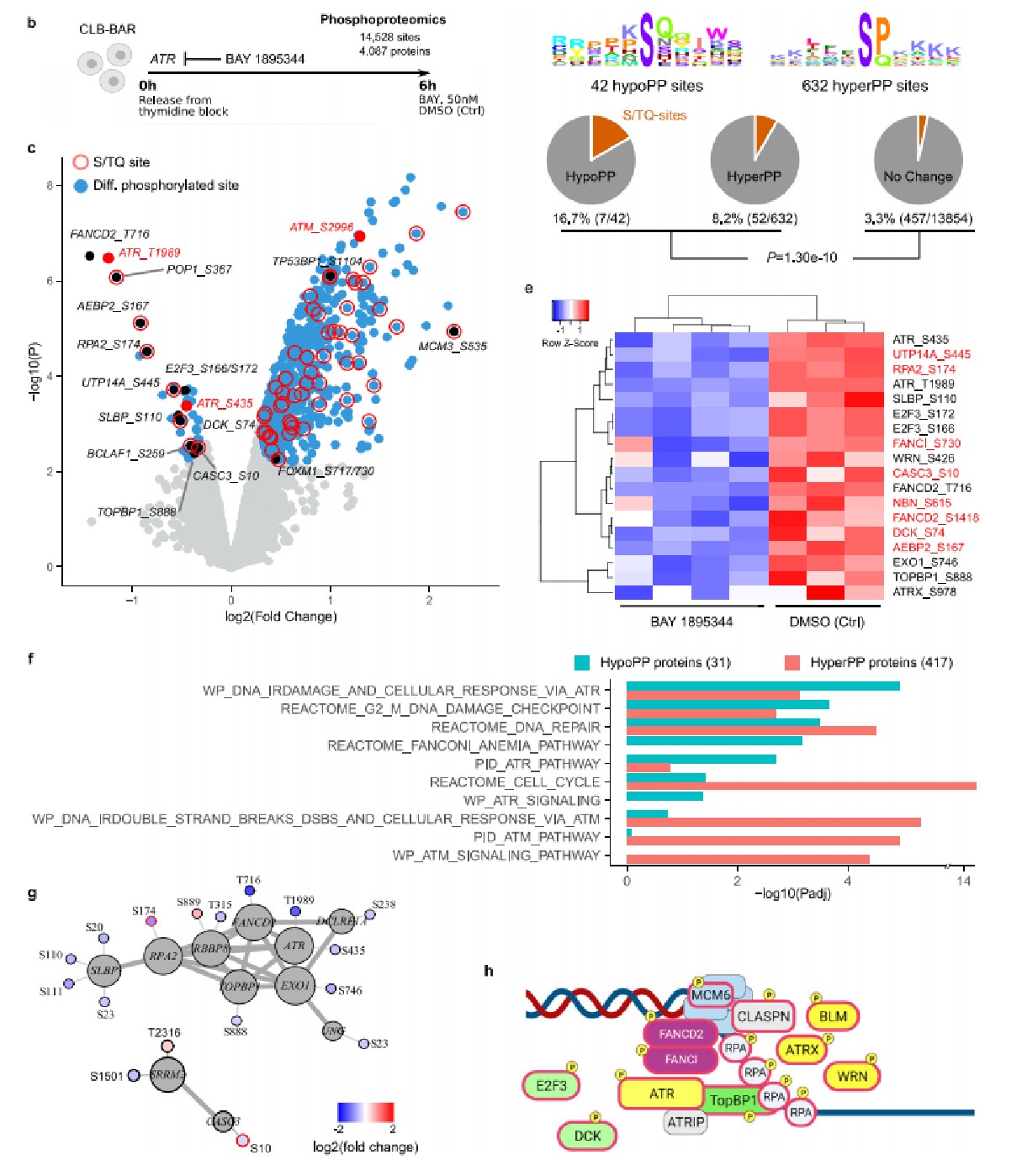
7. ATR抑制劑對NB細胞及小鼠移植瘤的影響
(1)ATR抑制劑(BAY1895344)抑制NB細胞增殖并誘導細胞死亡
BAY1895344和lorlatinib(一種ALK酪氨酸激酶抑制劑)單獨處理NB細胞顯著抑制了癌細胞的增殖,而BAY1895344和lorlatinib聯合治療能進一步抑制癌細胞增殖。
(2)BAY1895344抑制小鼠異種移植瘤的生長
BAY1895344和lorlatinib單獨或聯合治療CLB-BAR細胞的小鼠移植瘤模型均顯示出顯著的抑制移植瘤生長的作用。
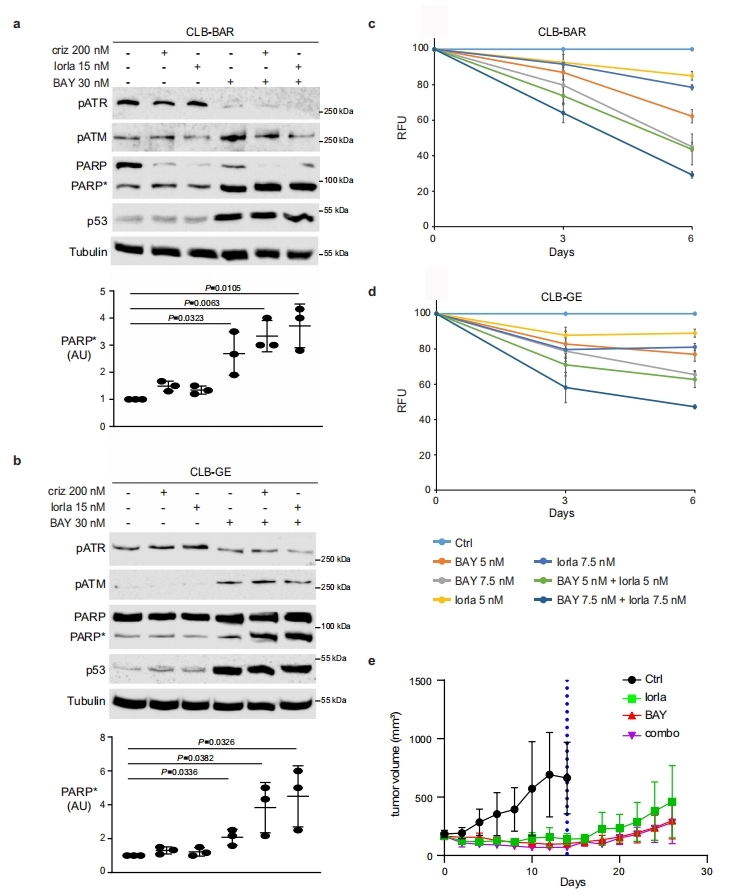
8. ATR抑制劑和ALK抑制劑聯合治療,消除了ALK驅動的NB基因工程小鼠的腫瘤
作者在兩個ALK驅動的NB基因工程小鼠模型(Rosa26_Alkal2;Th-MYCN (n = 4)和Alk-F1178S;Th-MYCN(n = 4))上檢測了BAY1895344和lorlatinib聯用的治療效果。14天的藥物聯用治療后,所有小鼠均未檢測到腫瘤,而對照組則有顯著的腫瘤生長。單獨的lorlatinib治療可以減少腫瘤增長,但并不能使腫瘤完全消退。在一個療程的藥物聯用治療結束后,作者繼續跟蹤了小鼠的腫瘤生長,通過超聲和MRI檢測確認在治療開始后的第50天,腫瘤也是完全消退的。在治療結束更久的時間后會發生腫瘤的復發,但繼續進行藥物聯用治療依然有抗腫瘤響應。對比藥物聯用和單獨使用lorlatinib可以確定,BAY1895344和lorlatinib聯用可使ALK驅動的NB腫瘤完全消退。
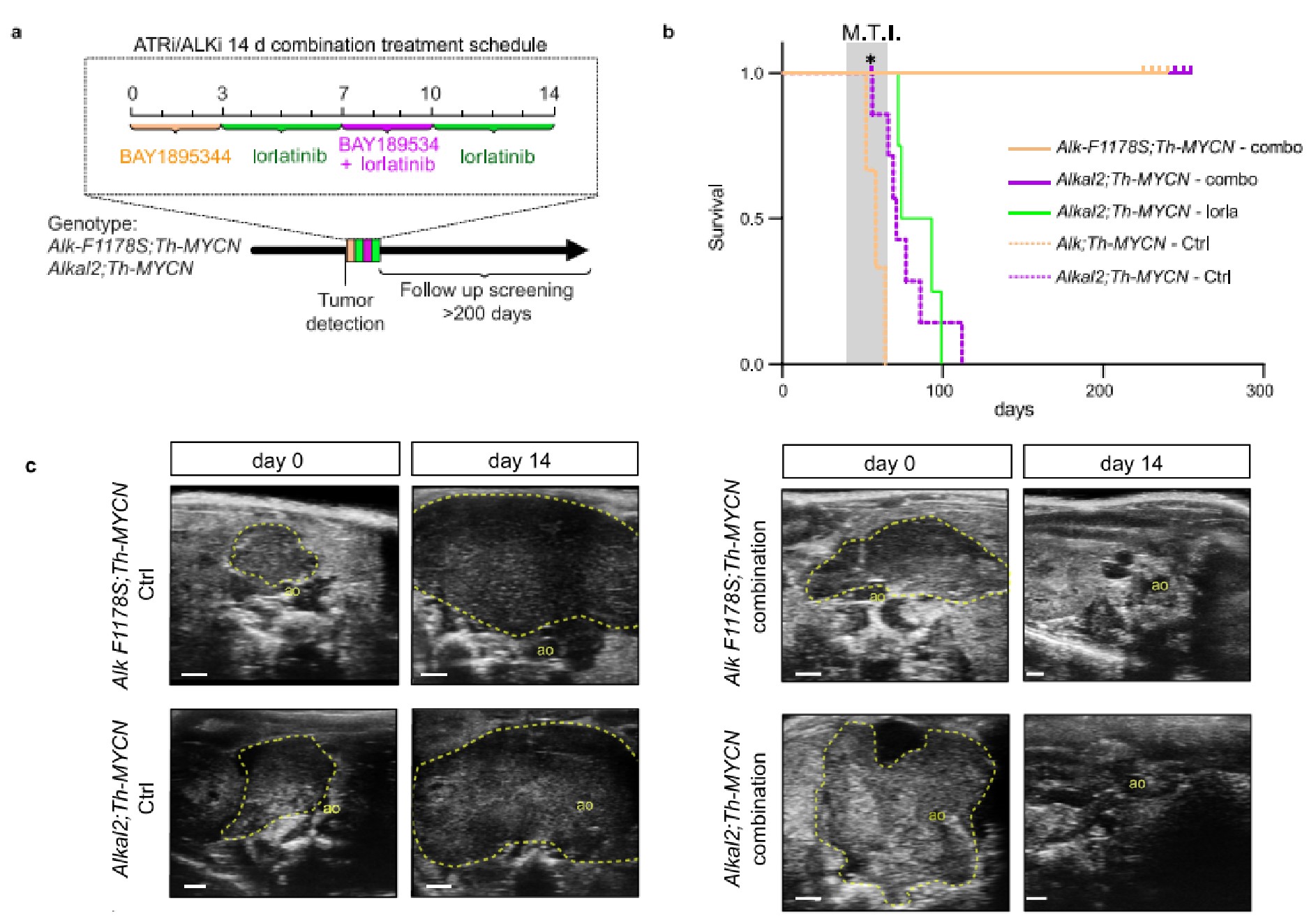
通過轉錄組分析ATR抑制劑(BAY1895344)治療ALK驅動的NB基因工程小鼠(Rosa26_Alkal2;Th-MYCN (n = 3)和Alk-F1178S;Th-MYCN(n = 3)))的作用機制。兩個不同小鼠模型RNA測序分別發現了603和2220個差異基因,這些差異基因與此前在NB細胞系中發現的差異基因大致相同,都顯示了E2F轉錄因子靶基因和G2M檢查點相關蛋白的富集。免疫組化也證實了BAY1895344治療后,腫瘤細胞凋亡增加、增殖減少。由于FOXM1是ATR的磷酸化靶點,作者觀察到在BAY1895344治療的腫瘤中,pFOXM1的亞細胞定位發生了顯著變化。治療前,pFOXM1定位于細胞核中,而在治療后,pFOXM1則定位于細胞質中。此外,作者還發現,BAY1895344治療誘導了強烈的免疫響應。
最后,GSEA分析發現的標志基因集與此前NB細胞系中發現的幾乎相同。如,與ATR表達正相關的基因顯著富集在E2F轉錄因子的細胞周期相關靶點和G2/M檢查點,與ATR表達負相關的基因富集在包括p53通路在內的其他通路。該結果也表明了ATR抑制造成的基因表達的變化在作者的模型中具有高特異性。
研究總結
本研究通過在細胞系、異種移植瘤模型以及基因工程小鼠模型中確認了ATR抑制劑BAY1895344是一種有效的神經母細胞瘤抑制劑。尤其是,14天的ATR和ALK抑制劑聯合治療方案完全消除了所有ALK驅動的NB基因工程小鼠的腫瘤。ATR抑制劑治療后的基因工程小鼠的磷酸化蛋白質組學和轉錄組分析發現,藥物治療引起了強烈的E2F轉錄因子的響應,該結果與細胞系上的轉錄組和蛋白組學發現一致。此外,研究還發現,ATR抑制劑抑制ATR后,伴隨著炎癥特征和廣泛的腫瘤免疫浸潤,這意味著ATR抑制不會對免疫反應產生負面影響。綜上所述,這些結果強烈表明抑制ATR對ALK驅動的神經母細胞瘤有有效的治療效果,具有潛在的臨床應用價值。
做蛋白組學· 找吉凱
吉凱基因憑借多年在靶標篩選及驗證服務領域的技術積累,建立的標準化 、工程化 、系統化的GRP平臺,為中國研究型醫生提供科研服務,加快科研成果轉化。其中,蛋白質組學平臺擁有多臺timsTOF Pro、Exploris 480高精度質譜儀,專業的Spectronaut Plusar、Mascot等分析軟件,提供專業的4D、DIA、TMT、PRM、磷酸化修飾組等檢測服務,強大的機器學習算法、IPA分析、蛋白基因組分析服務,系統的生物標志物、分子分型、藥物靶點、基因功能研究等解決方案,真正讓廣大研究型醫生的科研工作更省心、更省力、更高效。更多實驗服務請訪問我們的官網:www.genechem.com.cn
上海吉凱基因醫學科技股份有限公司
地址:上海張江高科技園區愛迪生路332號
郵編:201203
全國熱線:4006210302
售前顧問:18616237127;18616759206
郵箱:service@genechem.com.cn
官網:www.genechem.com.cn