罕見病,不止罕見!罕見病遠超人們的想象:已知罕見病超過7000種,我國公布的罕見病目錄中已納入207種疾病,涉及遺傳(超過150種)、免疫(如自身免疫性腦炎、系統性硬化癥等10余種)及腫瘤相關疾病;全球約3億罕見病患者,我國罕見病群體規模約2000萬人……近年來,斑馬魚因其與人類基因同源性高達87%、胚胎通體透明、基因編輯效率高、繁殖力強且適合高通量篩選等獨特優勢,正在讓罕見病研究突破想象的邊界!
本期,我們介紹幾種環特生物可以為客戶提供的基于斑馬魚構建的常用罕見病模型,包括Werner綜合征、先天性脊柱側凸、Acrofacial Dysostosis–Cincinnati 綜合征(辛辛那提型肢面發育不良,AFDCIN)、Dravet綜合征(DS)等,為罕見病藥物篩選、致病機制研究及基因治療探索提供科學的評價工具及科學依據,助力罕見病精準診斷,讓罕見被看見!歡迎有需要的客戶垂詢!
01、構建斑馬魚Werner綜合征模型
Werner綜合征(WS),是一種罕見的常染色體隱性遺傳病,表現為加速衰老,但Wrn敲除小鼠無法重現患者表型,缺乏理想的脊椎動物模型。
通過建立高效的脊椎動物早衰模型——Werner綜合征特征的斑馬魚突變體,利用該模型進行大規模化合物篩選,并在早衰模型、生理性衰老斑馬魚及人類細胞中驗證,最后闡明有效藥物延緩衰老的分子機制。
wrn基因突變體斑馬魚在幼魚期就表現出與Werner綜合征患者相似的早衰特征,可作為高效的抗衰藥物篩選平臺,并在此基礎上從小規模篩選中發現抗癌藥物sapanisertib具有延緩衰老的潛力。
研究方法及指標
1. 通過ENU誘變篩選,獲得wrn基因突變體(第5外顯子T→A突變,提前終止密碼子);
2. 細胞衰老標志:SA-β-Gal染色、p21/p53表達檢測;
3. DNA損傷:γH2AX免疫熒光;表觀遺傳改變;腸道屏障功能;炎癥反應;內質網應激;生存曲線、游泳活動度追蹤等。
示意圖展示
參考文獻:J. Ma,Y. Chen,J. Song,Q. Ruan,L. Li, & L. Luo, Establishment and application of a zebrafish model of Werner syndrome identifies sapanisertib as a potential antiaging drug, Proc. Natl. Acad. Sci. 2025
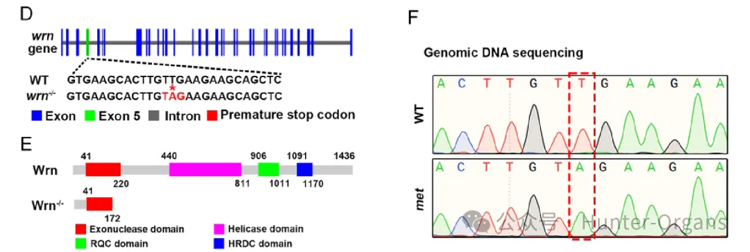
圖1
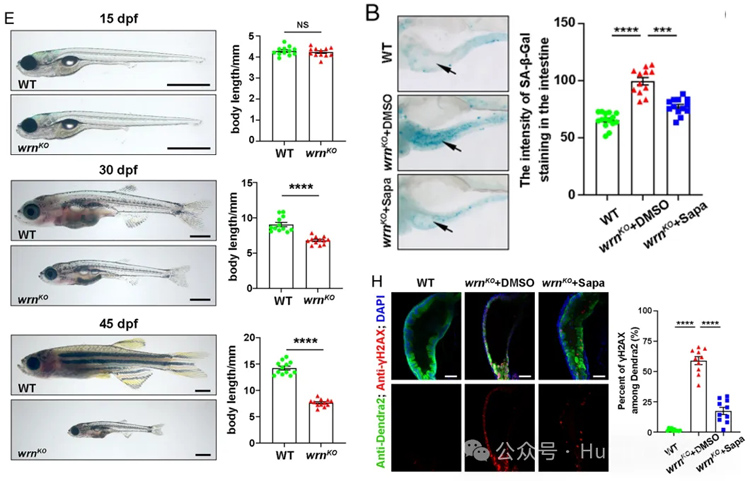
圖2
02、構建斑馬魚先天性脊柱側凸模型
先天性脊柱側彎,也稱先天性脊柱側凸(Congenital Scoliosis,CS),是由于胚胎在發育期間受各種原因(環境污染、基因突變、維生素缺乏、化學藥物因素以及病毒感染等)的影響,導致脊柱發育異常引起的脊柱畸形。
先天性脊柱側彎的發病率在活產嬰兒中約為(0.5~1)/1000,會影響嬰幼兒及青少年的骨骼發育,使身體變形,出現駝背、身材矮小扭曲的現象,嚴重者還可能影響心肺功能、腎臟功能、腸胃系統,心理等,甚至累及脊髓神經造成癱瘓。已發現Notch信號通路等多個基因突變可導致CS,但大多數家族性或散發性病例的遺傳原因仍未知。
通過ENU化學誘變篩選,鑒定導致先天性脊柱側凸樣椎體畸形的基因;解析分子機制,闡明該基因在脊柱發育中的具體作用機制,揭示該基因調控的下游信號通路及其與疾病的關系。
研究結果顯示,dstyk突變導致CS樣表型,突變體魚脊索液泡生物發生缺陷,導致軸骨分割模式破壞,成年期嚴重脊柱側凸和駝背、椎體畸形。脊索液泡是特化的溶酶體相關細胞器(LROs),Dstyk通過mTORC1/TFEB通路調控溶酶體/液泡生物發生。mTORC1抑制劑Torin1可部分rescue突變體表型,提示mTOR通路為潛在治療靶點。
研究方法及指標
1. 通過ENU誘變篩選,觀察體節縮短和脊柱彎曲表型,獲得smt(somite shorten)突變系,并定位到22號染色體dstyk基因;
2. 通過轉基因品系和活細胞成像、X-ray、micro-CT等連續觀察突變體魚脊索建立過程。
示意圖展示
參考文獻:Sun, X., Zhou, Y., Zhang, R. et al. Dstyk mutation leads to congenital scoliosis-like vertebral malformations in zebrafish via dysregulated mTORC1/TFEB pathway. Nat Commun 11, 479 (2020)
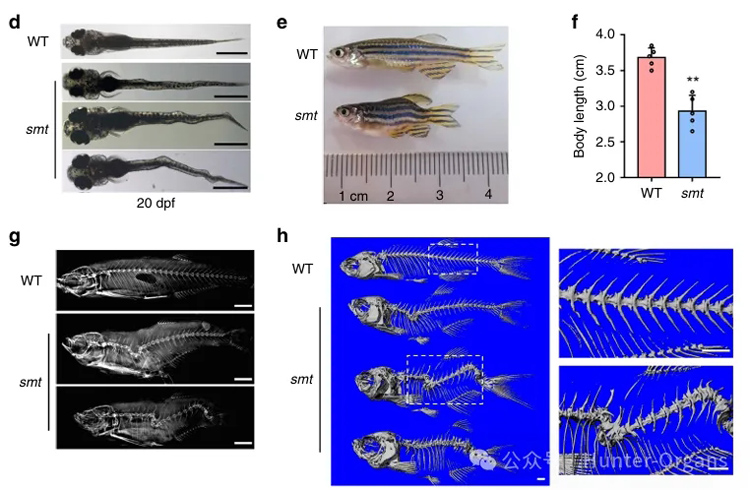
圖3
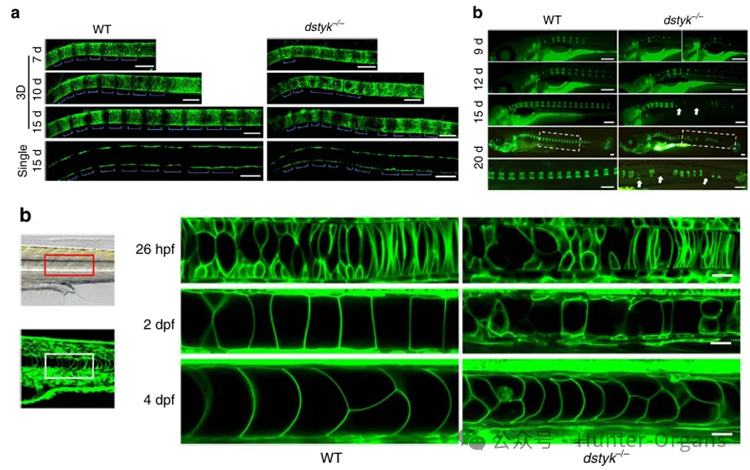
圖4
03、斑馬魚模型在AFDCIN綜合征中的應用
Acrofacial Dysostosis–Cincinnati 綜合征(辛辛那提型肢面發育不良,AFDCIN)是一種罕見的先天性顱面和肢體骨骼發育異常綜合征,表現為頜面發育不良和肢體畸形。該疾病由 POLR1A 基因的致病變異引起,POLR1A 編碼 RNA 聚合酶 I 的最大亞基,參與核糖體 RNA 的轉錄。
通過斑馬魚模型,研究 POLR1A 基因突變如何影響核糖體生物合成,進而導致顱面和肢體骨骼發育異常。研究 Tp53 信號通路在 AFDCIN 發病機制中的具體貢獻,并探索是否存在Tp53獨立的機制。
在 polr1a–/– 突變體中,Tp53 依賴的細胞凋亡是導致顱面骨骼發育不良的重要機制。通過遺傳抑制 Tp53(生成 polr1a–/–; tp53–/– 雙突變體),可以部分改善早期胚胎發育中的細胞凋亡和顱面骨骼發育不良,但無法完全恢復。
研究方法
通過基因編輯技術構建 POLR1A 基因敲除突變體(polr1a–/–)、 Tp53 突變體(tp53–/–) ,并生成雙突變體(polr1a–/–; tp53–/–)。
示意圖展示
參考文獻:Watt KEN, Neben CL, Hall S, et al. tp53-dependent and independent signaling underlies the pathogenesis and possible prevention of Acrofacial Dysostosis–Cincinnati type. Hum Mol Genet. 2018
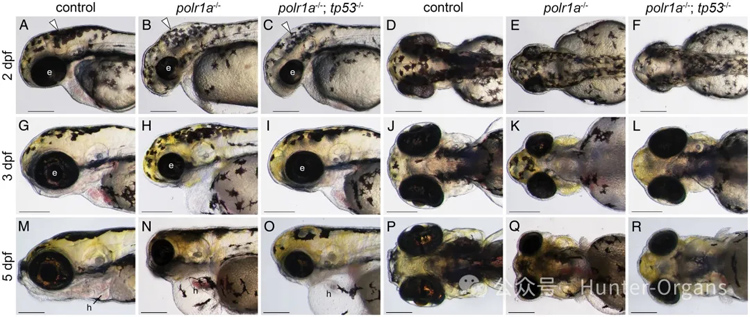
圖5
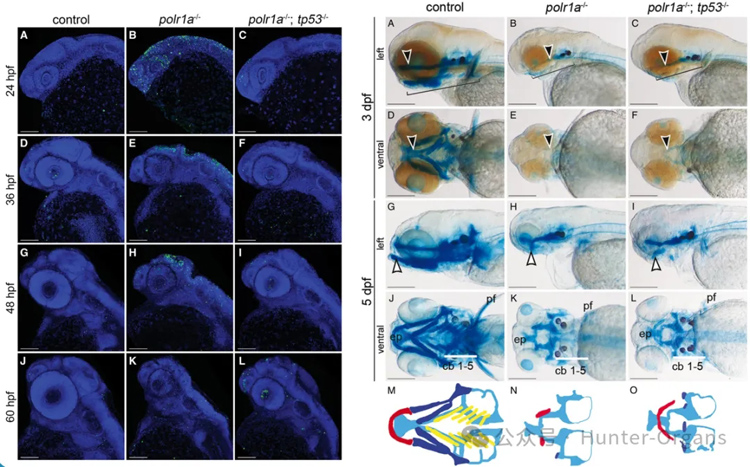
圖6
04、斑馬魚模型在嬰兒嚴重肌陣攣性癲癇中的應用
Dravet綜合征(DS)是一種由SCN1A基因突變引起的嚴重兒童期癲癇,其特征包括智力障礙、社交發展受損和持續的藥物難治性癲癇發作。SCN1A基因編碼的Nav1.1是一種電壓門控鈉通道,其突變與DS的發生密切相關。目前,DS的治療選擇有限,且大多數抗癲癇藥物(AEDs)對DS患者的療效不佳。
通過斑馬魚模型,篩選能夠改善DS相關癲癇表型的化合物,以期發現新的治療DS的藥物。scn1Lab突變體斑馬魚表現出與DS患者相似的癲癇表型,包括自發的癲癇發作、過度活躍和驚厥行為。通過高通量藥物篩選,研究團隊發現克列咪唑(clemizole)能夠顯著抑制scn1Lab突變體的癲癇發作行為和電圖癲癇活動。
研究方法
1. 利用化學誘變篩選得到了SCN1A基因突變的斑馬魚(scn1Lab),這些突變體表現出自發的異常電圖活動、過度活躍和驚厥行為;
2. 設計了一個高通量藥物篩選策略,通過監測斑馬魚的游泳行為和電圖活動,篩選能夠抑制突變體癲癇發作的化合物。
結果展示
參考文獻:Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410.
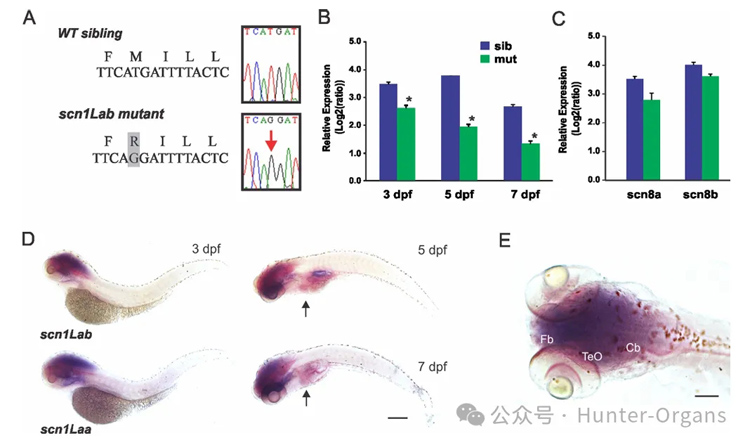
圖7
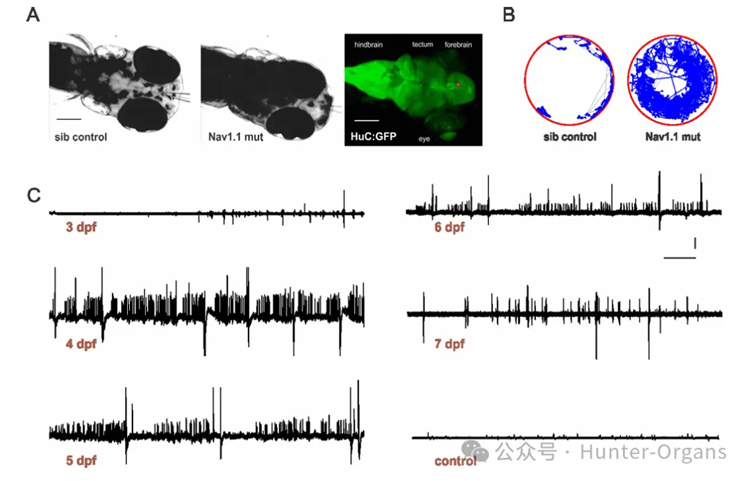
圖8