編者按
在生命科學的戰場上,每一次突破都可能改變無數家庭的命運。無論是兒童白血病、先天性心臟病,還是Werner綜合征、先天性脊柱側凸、Acrofacial Dysostosis–Cincinnati 綜合征(辛辛那提型肢面發育不良,AFDCIN)、Dravet綜合征(DS)……超過7000種罕見病,超過150種遺傳性疾病,長期以來都是醫學界難啃的“硬骨頭”。致病基因如何確認?疾病機制如何解析?潛在藥物如何快速篩選?
環特生物依托斑馬魚、類器官、基因編輯三大核心技術有機串聯,構建了一系列常用罕見病模型,形成了“基因編輯建模 → 斑馬魚初篩 → 類器官驗證”的全鏈條研究體系,為罕見病藥物篩選、致病機制研究及基因治療探索提供科學的評價工具及科學依據,打通科研到臨床的“最后一公里”!歡迎有需要的客戶垂詢!

案例一:用于兒科血液系統疾病研究
急性淋巴細胞白血病是一種常見的侵襲性兒童血液癌癥,在臨床急性淋巴細胞白血病患者中發現IL7R存在功能獲得型突變,但是該突變能否驅動白血病的發生,以及它是否與其它癌基因在白血病中有協同作用仍不清楚。
利用斑馬魚模型中的轉基因和細胞移植等方法,該研究揭示單獨的IL7R功能獲得型突變可誘導T細胞急性淋巴細胞白血病,進一步機制研究發現IL7R與MYC對于T細胞急性淋巴細胞白血病的發生有協同作用,且IL7R通路的激活增加了MYC誘導的T細胞急性淋巴細胞的擴散。該研究表明斑馬魚模型不僅能快速驗證急性淋巴細胞白血病相關的致病基因,也為兒科白血病聯合靶向治療提供了機制依據。
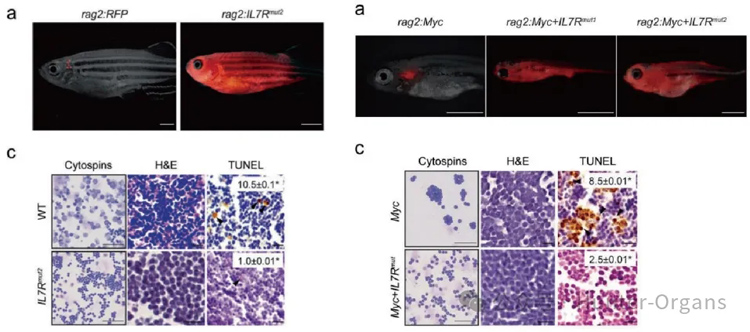
來源:Oliveira et al., 2022, Leukemia
案例二:用于兒科先天性心臟缺陷研究
左心發育不良綜合征(HLHS) 是嚴重先天性心臟病,特征為左心系統(心室、瓣膜、主動脈)發育不全,此前認為是多基因協同導致的表型疊加疾病。本研究構建了Rbfox1和Rbfox2雙敲除(DKO) 斑馬魚模型,其核心表型與人類HLHS 高度重疊:
①結構層面:心室發育不良、心臟瓣膜異常、主動脈(流出道)狹窄,與人HLHS 的左心系統發育不全特征一致;
②功能層面:心輸出量、射血分數顯著下降,符合HLHS 患者的心臟泵血功能缺陷;
③外觀層面:心包水腫、血液淤積,模擬HLHS 患者的臨床并發癥表現,為HLHS 研究提供了可靠的在體模型。
心肌特異性過表達Rbfox、mRNA注射野生型人RBFOX2 均可挽救DKO 斑馬魚的缺陷表型,為HLHS 的靶向治療(如基因治療、剪接調節劑)提供潛在靶點。
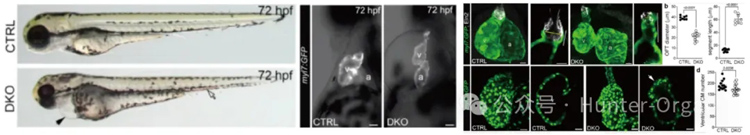
來源:Huang Met al.,2022,Nat Commun
案例三:兒科神經系統出生缺陷研究
神經管閉合失敗導致的NTDs是全球第二大常見出生缺陷,發病率約1/1000,主要包括無腦畸形、顱脊柱裂、開放性脊柱裂等。PAK2是Rho GTP酶的主要效應因子,調控細胞骨架網絡和多種生物學過程。PAK2功能缺失(敲除或突變)會激活BMP信號通路,導致細胞增殖減少、凋亡增加、分化軌跡異常,最終引發NTDs。
本研究構建斑馬魚突變體,該突變體在48 hpf時出現明顯神經管缺陷:前背側顱區存在腔隙、第三腦室和間腦發育異常,同時伴隨腦部出血,直接復刻了人類NTDs(如顱脊柱裂)和小鼠Pak2胚胎的核心表型。該模型證明PAK2同源基因(pak2a)的功能缺失會直接導致神經管閉合異常,驗證了PAK2在脊椎動物中調控神經管發育的保守性。斑馬魚作為易操作、發育快速的脊椎動物模型,既復刻了NTDs關鍵表型,又直接驗證了人類PAK2突變的致病性,同時強化了PAK2功能的跨物種保守性,為后續NTDs的機制研究和治療探索提供了可靠模型。
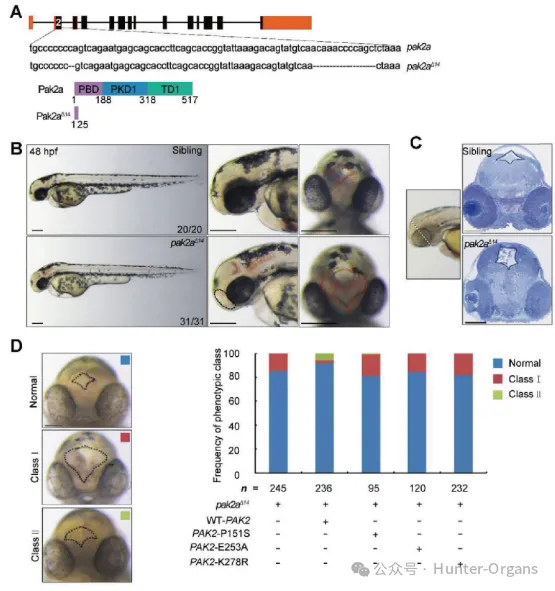
來源:WangY et al.,2023,Adv Sci(Weinh)
案例四:兒科先天性畸形疾病研究
兒科先天性畸形,即兒童出生時存在身體結構、功能或代謝異常,可由基因遺傳、環境或兩者交互作用引起。本研究在21個無關家庭的26名患有神經發育差異和多種先天性異常的個體中,發現了MAP4K4 基因的一系列罕見變異(包括8個截短變異、9個錯義變異和2個內含子剪接位點變異)。
這些個體表型與RASopathies綜合征重疊;通過斑馬魚模型驗證,這些變異分別表現為功能缺失(LOF)、顯性負性(DN) 或亞效等位基因效應,且MAP4K4在早期胚胎中作為RAS信號通路的負調控因子發揮作用,其功能異常(無論是增強還是減弱)都會導致斑馬魚出現顱面畸形、心臟缺陷等發育異常;同時,人類MAP4K4功能缺失可釋放對RAS信號的抑制,確立了MAP4K4作為綜合征性神經發育差異的致病基因,為相關疾病的分子診斷和治療提供了新靶點。
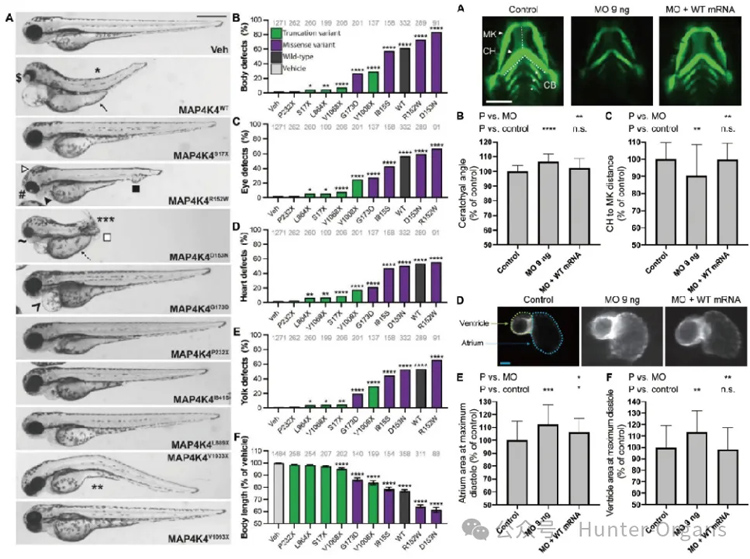
來源:Patterson et al.,2023,Sci.Adv
案例五:罕見遺傳腦病研究
無腦回畸形(也叫平滑腦),是一種罕見的遺傳異質性先天性腦畸形病,患者大腦表面平滑,且大腦皮層更厚,完全性無腦回畸形患者通常在2歲前死亡,不完全性無腦回畸形患者可長期存活,但通常伴有癲癇和智力殘疾。然而,這類疾病發病的分子機制仍不清楚。
2025年1月1日,耶魯大學張策等研究團隊在Nature發表最新研究,利用兩種不同類型的無腦回畸形患者細胞構建的無腦回畸形類器官,證實了mTOR 通路活性低下是無腦回畸形的臨床相關分子機制,拓展了對mTOR通路的認識,凸顯了大腦健康發育所必須達到的微妙平衡。
來源:nature/s41586-024-08341-9
案例六:利氏綜合征藥物開發與研究
利氏綜合征(Leigh Syndrome, LS),是一種極其嚴重的罕見病,它會剝奪孩童的運動能力,讓他們無法獨立行走,同時語言甚至是自主呼吸的能力也會受損,最終導致患兒走向生命終點。
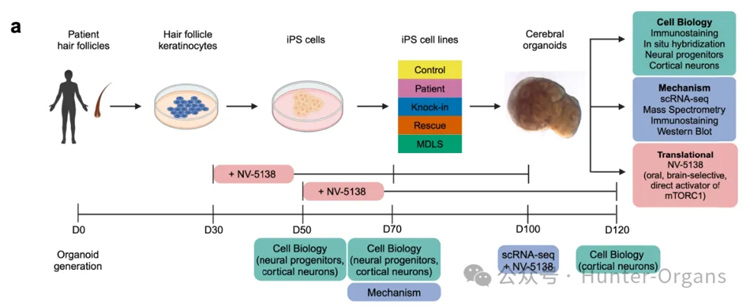
2026年3月19日,頂尖學術期刊《Cell》發表了一項具有里程碑意義的研究,依托患者來源的誘導多能干細胞(iPSCs),創新性地構建了神經前體細胞(NPCs)和三維(3D)皮質大腦類器官模型,不僅為利氏綜合征找到了首個具有明確臨床效果的候選藥物,更建立了一套可復制的“患者iPSC-類器官篩選-動物驗證-臨床轉化”的罕見病藥物研發路徑,解決了此前線粒體病尤其是mtDNA突變相關線粒體病難以構建疾病模型的核心問題,為后續大規模臨床試驗奠定了堅實基礎,也給全球無數無藥可治的線粒體病患者帶來了新希望。
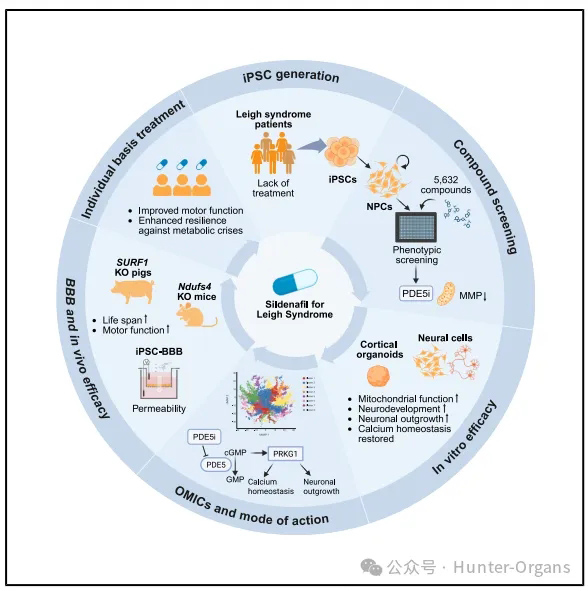
來源:Cell (2026). DOI: 10.1016/ j.cell.2026.02.008
每一次基因編輯的精準靶向,每一條斑馬魚的發育追蹤,每一個類器官的功能復刻,都是對遺傳性疾病研究的持續突破。環特生物以科技為刃,以循證為基,以斑馬魚、類器官、基因編輯三大技術,搭建起遺傳性疾病從基礎研究到臨床轉化的橋梁,為守護人類健康、攻克遺傳性疾病難題注入持久創新動力。
作為健康美麗產業CRO服務開拓者與引領者、斑馬魚生物技術的全球領導者,環特生物搭建了“斑馬魚、類器官、哺乳動物、人體”多維生物技術服務體系,開展健康美麗CRO服務、科研服務、智慧實驗室搭建三大業務。目前,環特已建立200多種斑馬魚模型,腦類器官、胃癌、心臟類器官及各種腫瘤類器官培養平臺,歡迎有需要的讀者垂詢!