點擊下文紅色鏈接,了解更多產品信息或撥打電話18521307698
摘要
代謝重編程是腫瘤放療抵抗的核心驅動機制,線粒體檸檬酸轉運蛋白 SLC25A1 是調控腫瘤代謝與放射敏感性的關鍵分子。本研究依托精準輻照儀(X?RAD 320)構建標準化肺癌細胞輻射模型,證實α?酮戊二酸(αKG)聯合 SLC25A1 特異性抑制劑 CTPI2 可通過誘導代謝重編程,顯著增強肺癌細胞放療敏感性。機制上,αKG 可促進 CTPI2 介導的致癌代謝物 D?2?HG 累積,加劇 DNA 損傷與氧化應激反應;煙酰胺(NAM)可通過調控 NAD+/NADH 氧化還原平衡逆轉上述效應。同時,賴氨酸去甲基化酶(KDMs)抑制劑 JIB04 可模擬 CTPI2 的放療增敏作用,且 αKG 可進一步增強其增敏效果。本研究為肺癌放療增敏提供了全新的代謝干預策略,精準輻照設備為代謝調控與放療抵抗機制驗證提供了核心技術支撐。

圖1:論文封面概要
材料與方法
本研究選用 NCI?H460、A549 人肺癌細胞系為研究對象,設置空白對照組、αKG 單獨處理組、CTPI2 單獨處理組、αKG+CTPI2 聯合處理組,并設立 NAM 補救組與 JIB04 干預組。采用 D?2?HG 檢測試劑盒定量分析致癌代謝物水平;通過堿性彗星實驗、γ?H2AX 焦點免疫熒光檢測評估 DNA 損傷程度;利用流式細胞術檢測細胞活性氧(ROS)水平、凋亡率及細胞死亡方式;借助 Seahorse 線粒體代謝分析儀評估線粒體功能;通過克隆形成實驗檢測細胞長期存活能力;采用雞胚絨毛尿囊膜(CAM)模型驗證聯合治療的體內抑瘤效果。
輻照實驗方案
依托生物輻照儀(X?RAD 320)實施電離輻射,儀器參數設置為:320 kV、12.5 mA,配備 1.65 mm 鋁濾片,照射距離 50 cm,劑量率 3.71 Gy/min。細胞經相應藥物預處理后,給予 2、5、8 Gy 梯度劑量照射;照射后立即置于細胞培養箱繼續培養,按實驗預設時間點收集樣本并開展后續檢測。
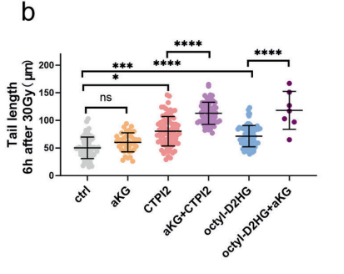
圖2:(原文 Fig.1B)適配輻照實驗方案,通過彗星實驗尾長量化輻射誘導的 DNA 損傷,印證模型構建有效性。
主要研究結果
1. αKG 協同 SLC25A1 抑制顯著增強放療增敏效應
αKG 聯合 CTPI2 可顯著促進 D?2?HG 累積,加劇輻射誘導的 DNA 雙鏈斷裂;在 CAM 體內模型中,聯合處理組腫瘤體積較單藥組顯著縮小,聯合放療后抑瘤效果進一步提升。
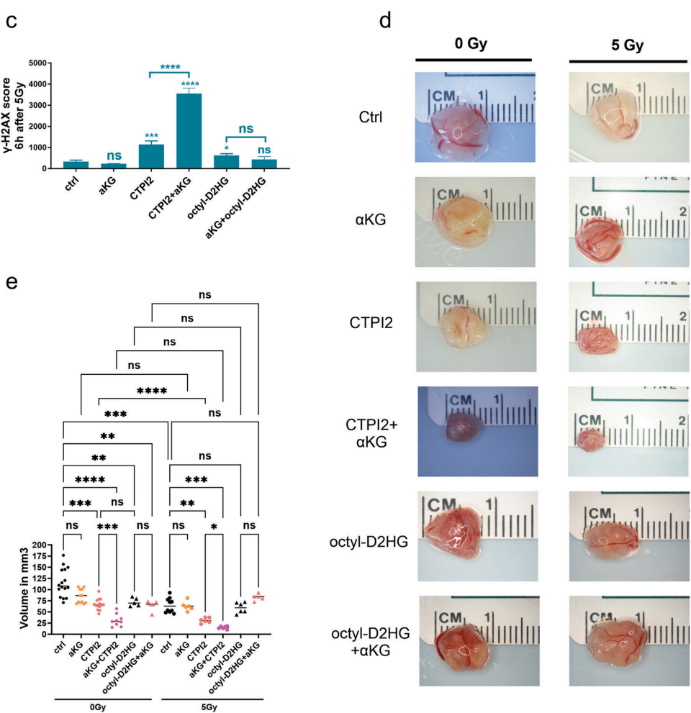
圖3:(原文 Fig.1C-E):量化 CAM 模型各組腫瘤體積,直觀呈現聯合放療的體內抑瘤效果
2. 代謝紊亂與氧化應激介導增敏作用
聯合處理可顯著升高細胞質與線粒體ROS 水平,抑制線粒體呼吸功能,擾亂 NAD+/NADH 氧化還原平衡,降低細胞增殖活力;NAM 預處理可有效逆轉上述代謝與氧化應激異常。
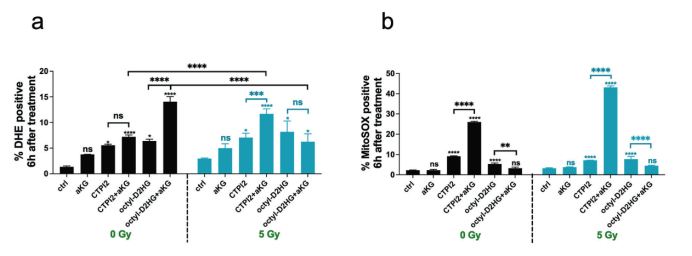
圖4:(原文 Fig.2A-B):聯合處理對胞質及線粒體 ROS 的提升作用,支撐氧化應激機制
3. KDMs 抑制與 αKG 聯用實現協同增敏
JIB04 單獨處理即可誘導 DNA 損傷與 ROS 生成,聯合 αKG 后,放療對細胞存活的抑制作用進一步增強,證實 KDMs 抑制是該代謝增敏策略的重要介導機制。
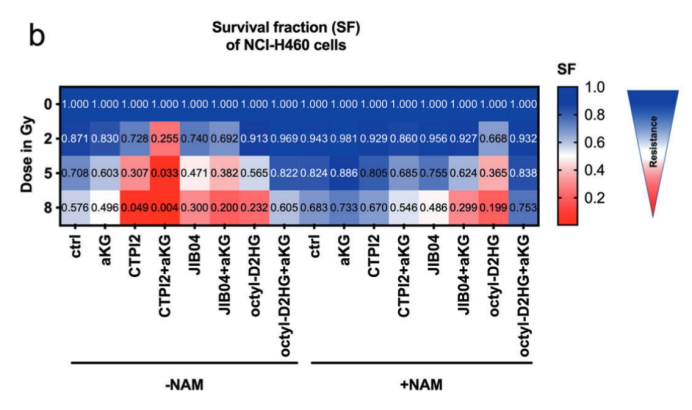
圖5:(原文 Fig.5B):通過存活分數熱圖,呈現不同處理組在梯度輻射劑量下的長期存活差異
結論
αKG 通過促進 D?2?HG 累積,協同 SLC25A1 抑制誘發 DNA 損傷、氧化應激與線粒體功能障礙,進而增強肺癌細胞放療敏感性,該效應依賴 NAD+/NADH 氧化還原平衡調控,且 KDMs 抑制是核心介導環節。X?RAD 320 輻照儀憑借精準劑量輸出、穩定照射條件與良好重復性,為代謝重編程介導的放療增敏機制研究、體內外療效驗證提供了可靠的標準化實驗平臺。αKG 聯合 SLC25A1 抑制或 KDMs 抑制,有望成為肺癌臨床放療增敏的新型代謝干預方案,為克服腫瘤放療抵抗提供全新思路與實驗依據。
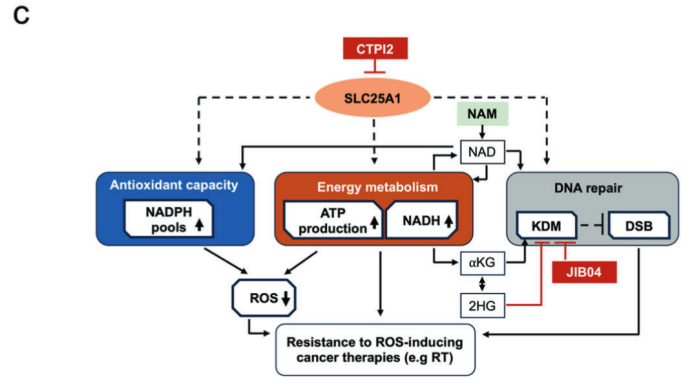
圖6:(原文 Fig.5C):SLC25A1 抑制聯合 αKG 通過代謝重編程增強放療敏感性的完整路徑示意圖
原文出處DOI:10.1038/s41420-024-01805-x