摘要
宮頸癌放療療效常受輻射抵抗限制,鐵死亡為提升放射敏感性提供新方向。本研究借助 X-RAD 320 ix 輻照儀構(gòu)建標準化輻射模型,揭示核心機制:HSP90 通過招募泛素連接酶 WWP1/2 抑制 NEDD4L,進而穩(wěn)定脫氧胞苷激酶(dCK);dCK 通過轉(zhuǎn)錄因子 ESR1 上調(diào) SLC7A11 表達,抑制輻射誘導的鐵死亡,導致宮頸癌抵抗。HSP90 抑制劑 17-AAG 可阻斷該通路,增強放療效果。這一發(fā)現(xiàn)為宮頸癌放療增敏提供新策略,也彰顯了專業(yè)輻照儀在腫瘤放療機制研究中的核心價值。
方法
選用宮頸癌 HeLa 細胞系,構(gòu)建 dCK、HSP90 敲低 / 過表達穩(wěn)定細胞系;采用 BALB/c 裸鼠建立皮下移植瘤模型,分為對照組、17-AAG 組、放療組、17-AAG + 放療組。通過 X-RAD 320 ix 輻照儀對細胞和裸鼠進行電離輻射,模擬臨床放療場景。結(jié)合 Western blot、免疫沉淀、流式細胞術(shù)、熒光染色、克隆形成實驗、RT-qPCR 及動物實驗,驗證蛋白相互作用、鐵死亡相關指標及放療增敏效果。
輻照儀的具體實驗方法
采用 X-RAD 320 ix 輻照儀進行細胞和動物電離輻射處理。細胞照射時將培養(yǎng)板置于指定位置確保均勻受照,設置 4 Gy、8 Gy 核心劑量,劑量率 300 cGy/min;動物照射用鉛模保護正常組織,按 8 Gy×3 次的分次放療方案精準遞送劑量。儀器自動調(diào)控 320 kV 電壓、12.5 mA 電流等參數(shù)保證劑量準確,照射后按預設時間點開展后續(xù)檢測。
輻照儀的重點實驗結(jié)果
輻照儀穩(wěn)定輸出梯度劑量,成功構(gòu)建 HeLa 細胞輻射抵抗模型,8 Gy 照射后 dCK、HSP90 蛋白表達呈時間依賴性升高;通過精準劑量控制,明確 dCK 或 HSP90 敲低組在 4-8 Gy 劑量范圍內(nèi)克隆形成率顯著降低,脂質(zhì) ROS 積累增加;模擬臨床分次放療方案,在裸鼠模型中成功驗證 17-AAG 聯(lián)合放療的協(xié)同抑瘤效果,為臨床轉(zhuǎn)化提供可靠數(shù)據(jù)。
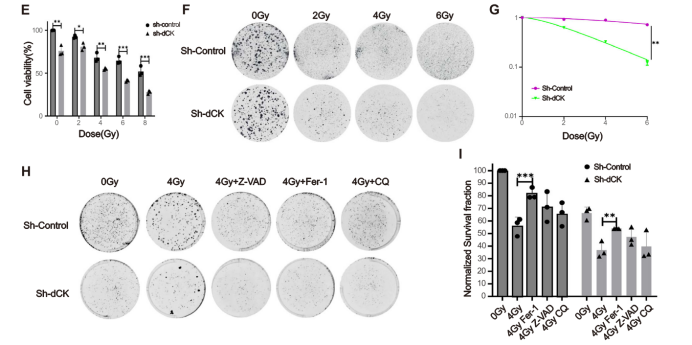
原文 Figure 1:呈現(xiàn)不同劑量輻射下 dCK 敲低組與對照組的克隆形成差異,輻照儀誘導的劑量依賴性放射敏感性差異
結(jié)果
dCK 在宮頸癌組織高表達且與不良預后相關,HeLa 細胞中 dCK 敲低可增強輻射誘導的鐵死亡,提升放射敏感性。dCK 與轉(zhuǎn)錄因子 ESR1 正相關,可激活 SLC7A11 啟動子活性,上調(diào)其表達并延長蛋白半衰期,減少脂質(zhì) ROS 和 Fe²?積累,抑制鐵死亡。HSP90 與 dCK 直接結(jié)合,招募 WWP1/2 降解 NEDD4L,抑制 dCK 泛素化降解,維持其蛋白穩(wěn)定性。HSP90 敲低或 17-AAG 處理可促進輻射誘導的鐵死亡,裸鼠實驗中 17-AAG 聯(lián)合放療顯著縮小腫瘤體積,且不影響小鼠體重。
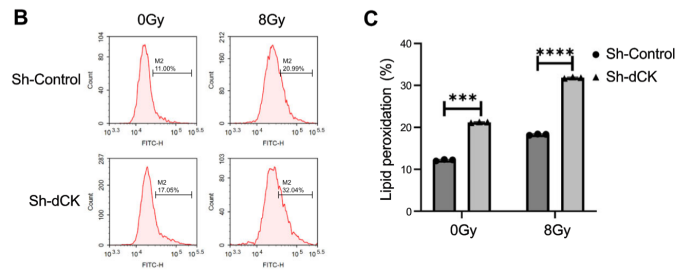
原文 Figure 2B-C:展示 dCK 敲低后輻射誘導的脂質(zhì) ROS 積累,直接支撐鐵死亡介導的增敏機制
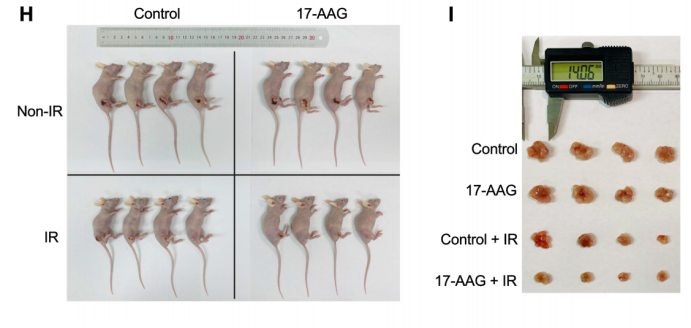
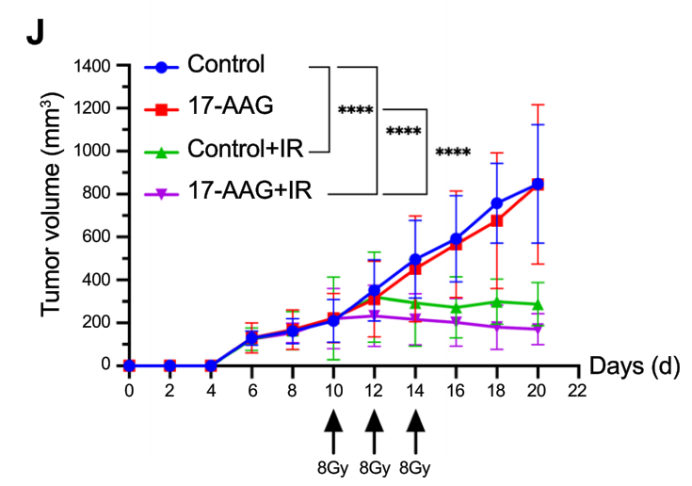
原文 Figure 6H-J:對比各組裸鼠腫瘤體積變化,明確 17-AAG 聯(lián)合放療的體內(nèi)協(xié)同療效
結(jié)論
HSP90-WWP1/2-NEDD4L-dCK-ESR1-SLC7A11 軸是調(diào)控宮頸癌輻射誘導鐵死亡的關鍵通路,HSP90 通過穩(wěn)定 dCK 抑制鐵死亡,導致放療抵抗。HSP90 抑制劑 17-AAG 可阻斷該通路,恢復腫瘤細胞鐵死亡敏感性,增強放療療效。X-RAD 320 ix 輻照儀憑借精準的劑量調(diào)控、穩(wěn)定的照射效果,為輻射模型構(gòu)建、敏感性量化及機制驗證提供核心工具,助力明確 HSP90/dCK 作為宮頸癌放療增敏靶點的潛力。該發(fā)現(xiàn)為克服宮頸癌放療抵抗提供新策略,也為腫瘤靶向放療研究提供新思路。
原文 Figure 8: HSP90/dCK 軸調(diào)控輻射誘導鐵死亡及 17-AAG 干預效果的完整路徑示意圖
①原文出處DOI:10.1038/s41420-025-02388-x