GDC-1971——SHP2抑制劑引領(lǐng)癌癥治療新紀元

簡介
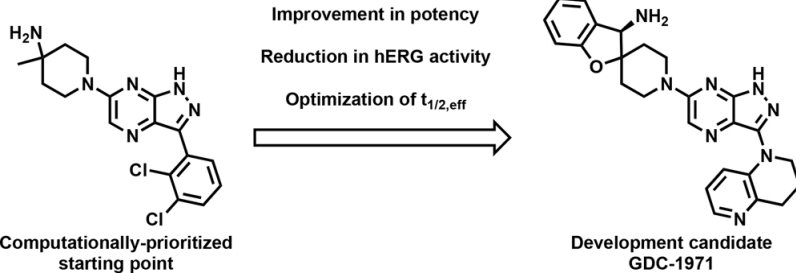
近年來,蛋白酪氨酸磷酸酶SHP2在介導RAS驅(qū)動的MAPK信號轉(zhuǎn)導過程中,已成為腫瘤學領(lǐng)域備受矚目的研究靶點。其治療方法不僅限于單藥使用,還能與KRAS抑制劑共同協(xié)作,展現(xiàn)出良好的治療潛力。研究者初步發(fā)現(xiàn),某些類似藥物能夠與異構(gòu)位點結(jié)合,進而使酶保持在封閉、非活性的狀態(tài)。特別值得一提的是,GDC-1971(原名RLY-1971)作為一種SHP2抑制劑,目前正在與KRAS G12C抑制劑divarasib(GDC-6036)聯(lián)合應(yīng)用于臨床試驗中,共同對抗由KRAS G12C突變驅(qū)動的實體瘤,為腫瘤治療領(lǐng)域帶來了新的希望。
GDC-1971的合成
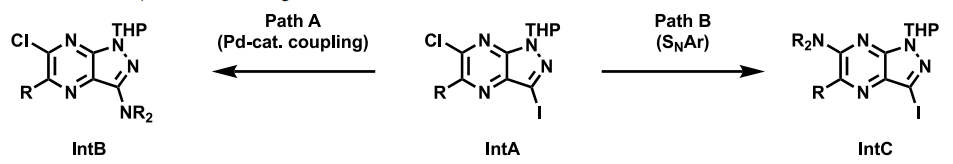
在藥物化學的優(yōu)化歷程中,GDC-1971等化合物主要遵循兩條合成路徑。這兩條路徑均可采用相似的化學技術(shù),針對核心結(jié)構(gòu)、堿性胺和四氫萘啶區(qū)域進行調(diào)整,進而將多樣化的結(jié)構(gòu)單元有效結(jié)合,從而實現(xiàn)化合物的優(yōu)化與改良。
類似物的一般合成
從2,6-二氯吡嗪起始,可以通過對其進行石化處理,并使用甲酸乙酯進行淬滅反應(yīng),從而合成得到3,5-二氯吡嗪-2-甲醛。鑒于這種醛的化學性質(zhì)較為不穩(wěn)定,為了更有效地分離它,傾向于將其轉(zhuǎn)化為相應(yīng)的亞硫酸氫鈉加合物形式。進一步地,當將此加合物與肼進行處理時,會生成一種加合物混合物。然而,在這過程中,相應(yīng)的腙會發(fā)生分子內(nèi)的環(huán)化反應(yīng),進而轉(zhuǎn)化為6-氯-1H-吡唑并[3,4-b]吡嗪。隨后,通過碘化過程,獲得了化合物44。為了得到所需的氯碘中間體45,接著采用二氫吡喃進行保護處理。
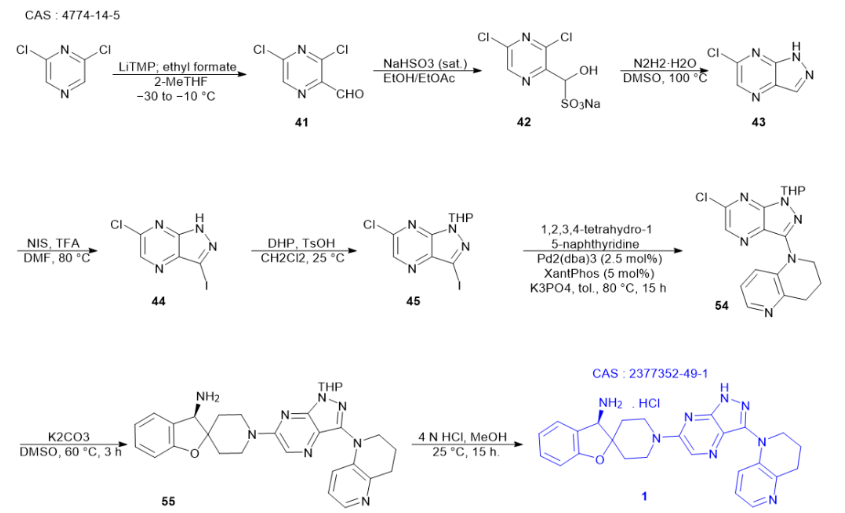
在堿性環(huán)境下,關(guān)鍵中間體1-(6-氯-1-(四氫-2H-吡喃-2-基)-1H-吡唑并[3,4-b]吡嗪-3-基)-1,2,3,4-四氫-1經(jīng)過一系列反應(yīng),首先與四氫萘啶和Xantphos通過鈀催化偶聯(lián)作用,生成了5-萘啶。隨后,這個雜芳基氯在二甲基甲酰胺中與(R)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺的雙鹽酸鹽和二異丙基乙胺發(fā)生親核芳香取代反應(yīng),最終得到GDC-1971的四氫吡喃保護類似物。為了進一步處理,這個鹽可以用氫氧化鈉水溶液進行游離,隨后用2-甲基四氫呋喃進行萃取。這一系列步驟精細而關(guān)鍵,為GDC-1971的制備提供了重要的中間體。

芳基螺旋環(huán)胺構(gòu)建塊的代表性合成
以下方案詳細描述了用于制備類似物的芳基螺環(huán)胺結(jié)構(gòu)單元的合成過程。在這一過程中,首先利用埃爾曼助劑進行不對稱還原反應(yīng),隨后進行全局脫保護步驟,從而得到二胺鹽。這一產(chǎn)物可以直接應(yīng)用于如IntA等化合物的親核芳香取代反應(yīng)中,為后續(xù)的合成步驟提供了關(guān)鍵的原料。

GDC-1971中螺苯并呋喃哌啶環(huán)的合成
(R)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺,作為GDC-1971的關(guān)鍵堿性胺成分,其合成起始于2-氟苯甲醛。隨后,通過一系列轉(zhuǎn)化步驟,成功將其轉(zhuǎn)化為相應(yīng)的1,3-二噻烷,為后續(xù)的合成反應(yīng)奠定了堅實基礎(chǔ)。
GDC-1971的藥理研究
GDC-1971,即(R)-1′-(3-(3,4-二氫-1,5-萘啶-1(2H)-基)-1H-吡唑并[3,4-b]吡嗪-6-基)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺,在生化及細胞環(huán)境中均展現(xiàn)出了對SHP2的顯著抑制效果。

GDC-1971的選擇參數(shù):結(jié)構(gòu)、理化和效價
為了更精準地預測GDC-1971在人體內(nèi)的藥代動力學(PK)特性及有效劑量,在臨床前物種中進行了PK評估,并收集了臨床前物種及人體系統(tǒng)中的體外ADME數(shù)據(jù),以提供全面的藥物代謝和動力學信息。

GDC-1971的多物種藥動學特征
GDC-1971的體外與體內(nèi)藥效、藥代動力學(PK)以及非靶向活性數(shù)據(jù)均顯示,該藥物能夠在適宜的劑量范圍內(nèi),以可接受的安全系數(shù)有效抑制人體內(nèi)的SHP2。目前,該化合物已順利進入進一步的安全性和IND使能研究階段,并有望最終進入臨床試驗。關(guān)于其后續(xù)的研究進展和臨床試驗結(jié)果,將在適當時機予以披露。

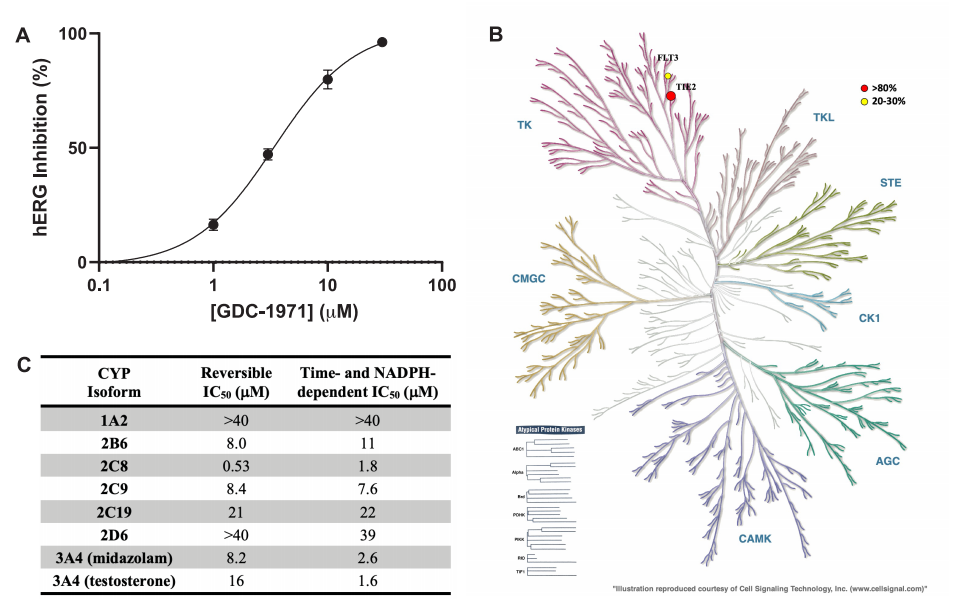
GDC-1971脫靶譜:(A)功能性hERG活性的劑量-反應(yīng)曲線。(B)樹突圖顯示一組激酶受到抑制。(C) CYP抑制譜。
結(jié)語
綜上所述,GDC-1971在體外和體內(nèi)臨床前階段所展現(xiàn)的特性,使其具備成為測試SHP2抑制作用的臨床試驗候選藥物的巨大潛力。依據(jù)本文詳述的合成路線,GDC-1971能夠利用批量制備的構(gòu)建模塊實現(xiàn)大規(guī)模合成,為后續(xù)的臨床研究提供了堅實的基礎(chǔ)。
參考文獻
1. Alexander M. Taylor. et al. Identification of GDC-1971 (RLY-1971), a SHP2 Inhibitor Designed for the Treatment of Solid Tumors. J. Med. Chem. 2023, 66, 19, 13384-13399. https://doi.org/10.1021/acs.jmedchem.3c00483