nat microbiol|全球腸道微生物群揭示腸桿菌科的雙面性:是威脅還是共生?

英文標題:Ecological dynamics of Enterobacteriaceae in the human gut microbiome across global populations
中文標題:全球人群腸道微生物群中腸桿菌科的生態動態
發表期刊:nature microbiology
影響因子:20.5
研究背景
人體腸道微生物群對健康意義重大,其失衡與多種疾病相關。腸桿菌科細菌是常見的機會致病菌,在健康人腸道中也普遍存在,提示種間相互作用可能影響感染抗性。但現有對腸桿菌科的研究多聚焦臨床分離株,且以往探究其與腸道微生物群關系的研究受樣本量和技術限制。如今宏基因組學發展提供了新契機,本研究利用大規模宏基因組數據,旨在揭示全球人群腸道微生物群中腸桿菌科的生態動態,為相關疾病防治和非抗生素療法開發提供依據。
研究結果
1、腸桿菌科的全球分布
為全面表征與腸桿菌科定植相關的人類腸道微生物群特征,研究者從45個國家的(歐洲35%、北美27.5%、亞洲23.2%、非洲8.4%)65項研究中檢索了12,238份公開的人類腸道宏基因組樣本(圖1)。基于統一人類胃腸道基因組(UHGG)目錄,準確檢測了全球宏基因組中4,612種腸道微生物(113種屬于腸桿菌科)的存在和豐度。
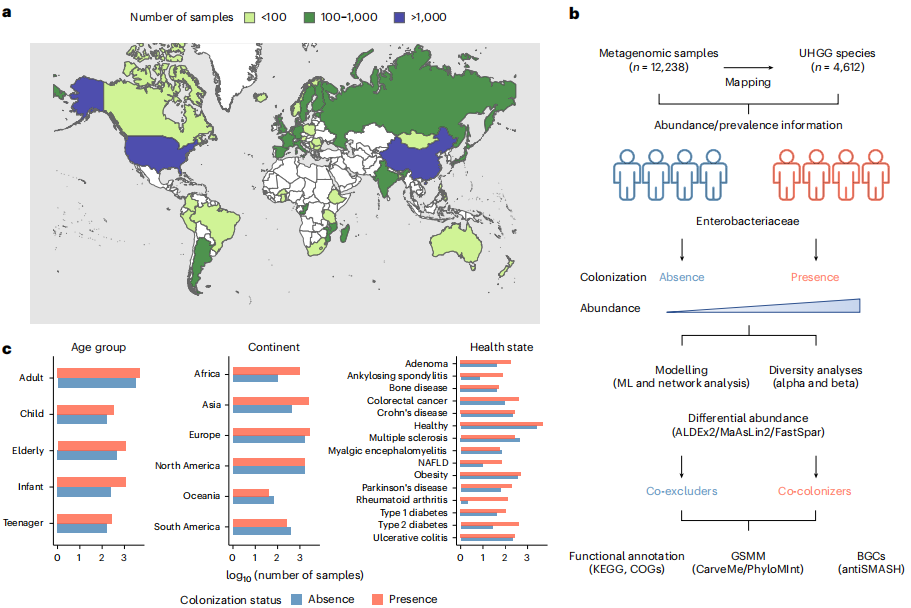
圖1. 探索腸桿菌科的全球生態景觀
研究發現,腸桿菌科的總體患病率為66%,其分布受地理、宿主狀態顯著影響:非洲樣本中大腸桿菌患病率最高(88%),嬰兒(74%)及類風濕性關節炎患者(96%)顯著富集(圖2a)。研究者進一步分析了腸桿菌科物種在腸道宏基因組中的共分布模式(圖2b),發現亞洲樣本中大腸桿菌與肺炎克雷伯菌共定植率達16.2%(P<0.001)。此外,在非洲和大洋洲樣本中,大腸桿菌、肺炎克雷伯菌和霍氏腸桿菌顯著共現(觀察比例5.3%vs預期比例0.7%)。總體而言,這些腸桿菌科共定植的地理差異可能反映了環境條件、飲食習慣、生活方式和醫療實踐的差異。研究通過宏基因組多位點序列分型(MLST)分析5,128份健康成人樣本,發現大腸桿菌存在585種序列型(STs),其中76.5%為未知STs(如非洲高豐度的 ST100024/83),揭示全球未被充分研究的菌株多樣性。
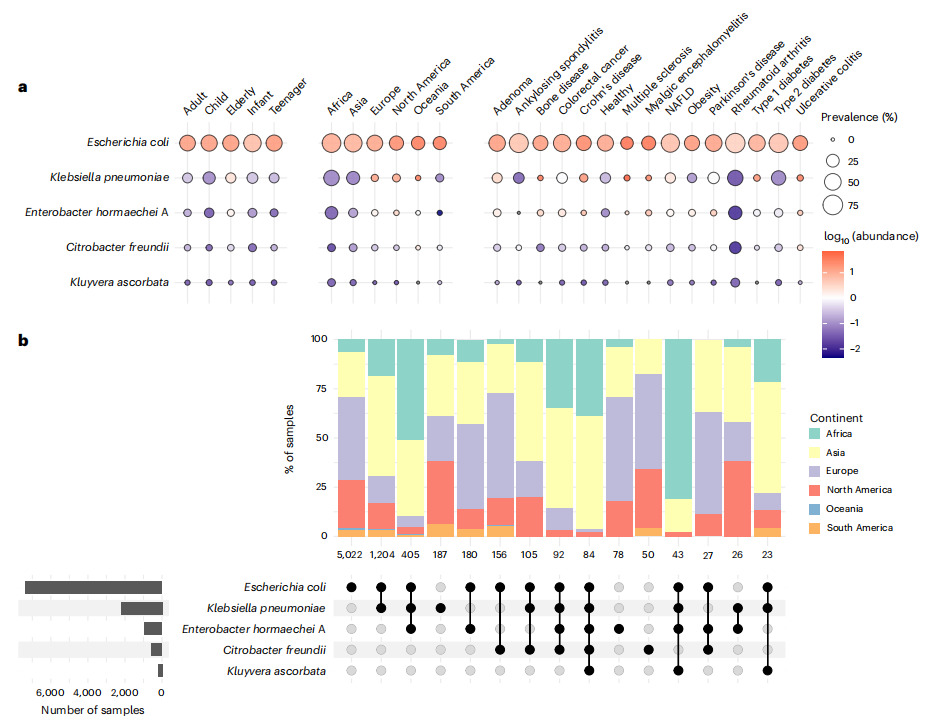
圖2. 最常見腸桿菌科物種的分布與多樣性
2、腸道微生物群結構與腸桿菌科定植動態的關聯
利用全球12,000份腸道宏基因組數據,研究者探索微生物群組成與腸桿菌科定植狀態的關系。基于非腸桿菌科物種的豐度和患病率,構建機器學習分類器來區分樣本是否含腸桿菌科(圖3a)。以腸桿菌科整體或大腸桿菌、肺炎克雷伯菌的定植狀態為因變量,測試三種監督學習方法,梯度提升法表現最優。
多樣性分析顯示,腸桿菌科陽性樣本的β多樣性顯著高于陰性樣本,且與α多樣性和測序深度無關。差異豐度分析結合ALDEx2和MaAsLin2模型,控制多種因素后,在眾多樣本中鑒定出與腸桿菌科等顯著相關的物種,分為共定植菌和共排斥菌(圖3b)。分類學特征顯示,毛螺菌目(Lachnospirales)、顫螺旋菌目(Oscillospirales)和擬桿菌目(Bacteroidales)多為共排斥菌,乳桿菌目(Lactobacillales)、韋榮球菌目(Veillonellales)和放線菌目(Actinomycetales)與共定植菌相關(圖3c)。
臨床相關性驗證結果表明,本研究中所鑒定出的共排斥菌/共定植菌,與針對產碳青霉烯酶腸桿菌科(CPE)的隊列研究中發現的差異物種之間存在顯著重疊。這一現象提示,這些物種或許并非僅對腸桿菌科的整體定植產生影響,而是有可能特異性地作用于CPE的定植過程。
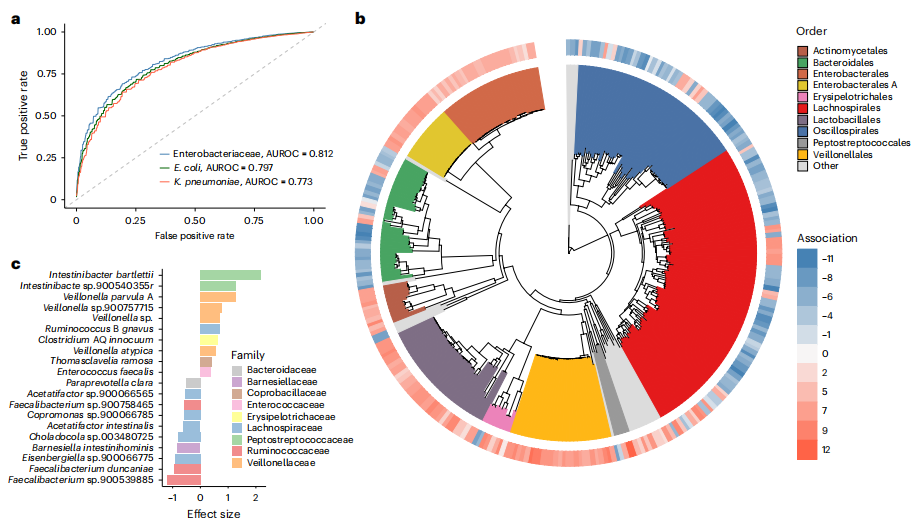
圖3. 腸道微生物組組成與腸桿菌科的定植和豐度有關
3、腸桿菌科定植的菌株特異性模式
上述分析揭示微生物組物種組成與腸桿菌科定植模式聯系緊密,然而菌株水平的差異也可能與腸桿菌科-微生物組之間的相互作用有關,研究者針對39種在健康成年人腸道內,與腸桿菌科存在共定植或共排斥關系的微生物展開研究。這些物種在統一人類胃腸道基因組(UHGG)中有充足高質量基因組代表。
借助統一人類胃腸道蛋白質(UHGP)目錄,研究者分析了這39個物種的附屬基因組,識別與腸桿菌科定植相關的亞種群體。結果發現部分附屬基因與腸桿菌科定植狀態顯著相關,且多富集于核苷酸轉運和代謝相關功能,主要涉及多形瘤胃球菌(Ruminococcus B gnavus)和福氏糞單胞菌(Faecalimonas phoceensis)。
研究者進一步研究了與腸桿菌科相關的附屬基因數量最多的菌株(前10%的菌株)的系統發育相似性。結果表明,與Ruminococcus B gnavus基因組相比,Faecalimonas phoceensis中與腸桿菌科共定植相關的種群結構要強得多(圖4)。在眾多候選物種中,只有2種糞單胞菌屬物種被確定為腸桿菌科的共定植物種。相較于其他相關菌屬,糞單胞菌屬的多樣性與腸桿菌科共定植之間存在更特定的菌株水平關聯。
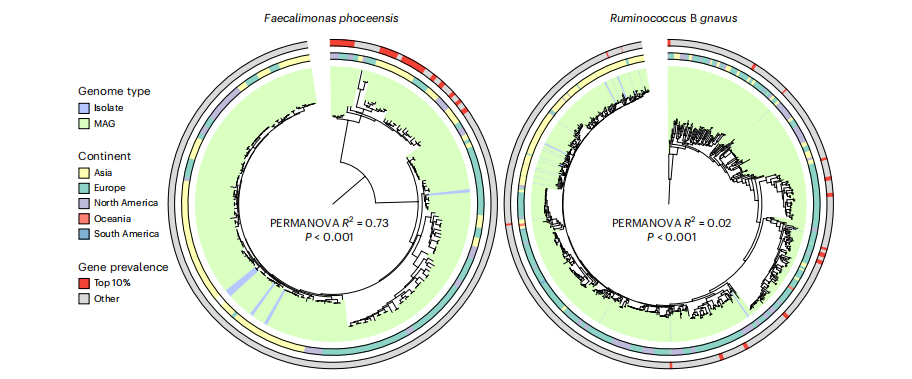
圖4. 福氏糞單胞菌呈現出菌株特異性的共定植模式
4、共定植物種功能多樣
作為分類學研究的補充,研究者分析307種物種中245種非腸桿菌科物種的功能和多樣性,這些物種在不同數據集和分類群中呈共定植或共排斥模式。基于KEGG注釋發現,共定植物種功能多樣性更高(圖5a),與注釋覆蓋范圍和基因組質量無關,與更高的代謝獨立性相關。統計分析KEGG直系同源基因(KOs)發現,共定植與耐藥性、DNA調控功能關聯強,共排斥與鐵代謝、轉運基因關聯強,暗示腸桿菌科和共排斥物種存在鐵競爭。研究者對與腸桿菌科共定植和共排斥相關的主要功能類別(直系同源聚類組,COGs)進行了特征描述(圖5b),共定植物種富含代謝相關功能(如氨基酸、核苷酸和無機離子代謝),支持其代謝獨立性更強的假設;共排斥物種編碼更多信號轉導相關基因(如孢子形成、運動性和群體感應),還有較多功能未知基因,或在抵抗定植中起作用,即使僅考慮芽孢桿菌門的物種(該門包含數量最多的共排斥物種和共定植物種),這些結果依然一致。利用gutSMASH算法研究發現,共排斥物種富含產短鏈脂肪酸及涉及 Rnf 復合物等的代謝基因簇(圖5c)。這強化了糞桿菌屬(腸道中顯著的短鏈脂肪酸生產者之一)的相關信號,也凸顯鐵在共排斥物種中的重要性。綜上,與腸桿菌科共存的物種功能多樣,鐵代謝和短鏈脂肪酸在調節腸道環境、調控腸桿菌科定植和豐度上或起重要作用。
研究者構建了候選共定植物種、共排斥物種及特定腸桿菌科物種的基因組規模代謝模型,計算代謝競爭和互補指數。結果顯示,競爭與互補指數呈負相關,共定植物種與腸桿菌科的代謝距離低于共排斥物(圖5d),表明生境篩選可能是腸桿菌科相關物種定植及微生物組組裝的主因。此外,組內代謝距離小于組間,且模擬不同飲食培養基時結果一致,佐證了營養需求相似的腸道微生物易在個體間共存的觀點 。
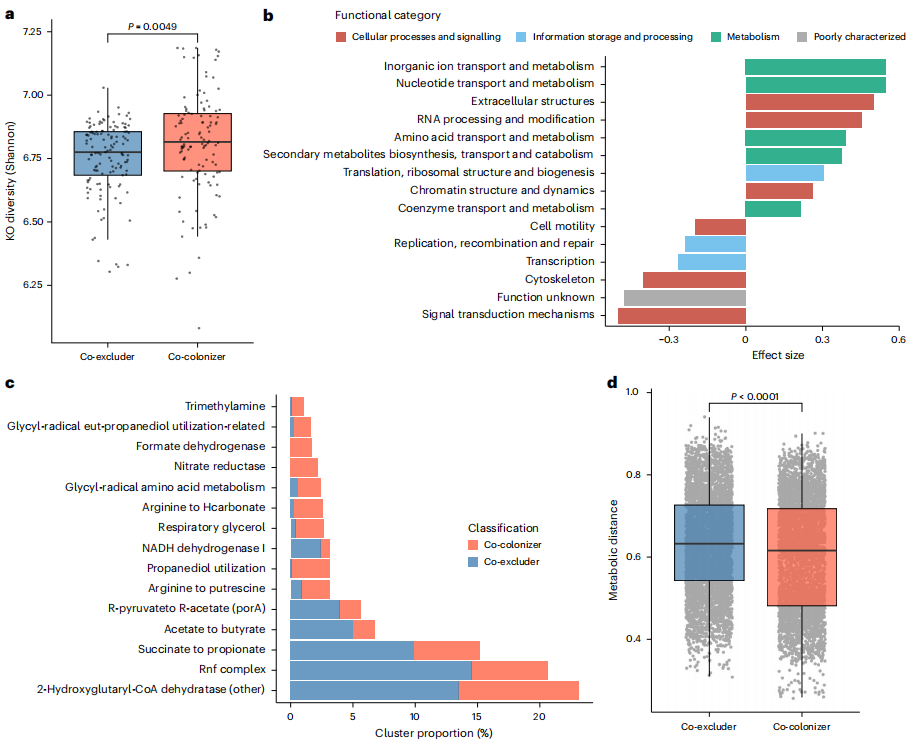
圖5. 共排斥物種和共定植物種之間的功能差異
5、共排斥物種編碼參與群體感應的基因
鑒于次級代謝產物影響細菌適存度與種間生態互作,研究者借助antiSMASH預測工具,分析腸桿菌科共定植物種和共排斥物種的生物合成基因簇(BGCs)。結果發現,共排斥物種顯著富集環狀內酯自體誘導物相關BGCs(圖6a)。自體誘導物作為群體感應信號分子,與COG分析中,共排斥物種信號轉導機制基因富集的結論相符。多數自體誘導物BGCs由毛螺菌科物種攜帶。按特定基因相似度分組(覆蓋度超過50%且核苷酸同一性超過50%),識別出獨特BGC家族,且這些家族在共定植物種和共排斥物種間存在差異(圖6b)。將家族基因與 “生物合成基因簇最低限度信息” 數據庫對比,發現與已知BGCs氨基酸同一性低(圖6c),表明它們多未被表征。因群體感應分子能助力腸道微生物抵御外來病原體定植,共排斥物種中自體誘導物BGCs的富集,可能調控腸桿菌科在人體腸道的定植和豐度。
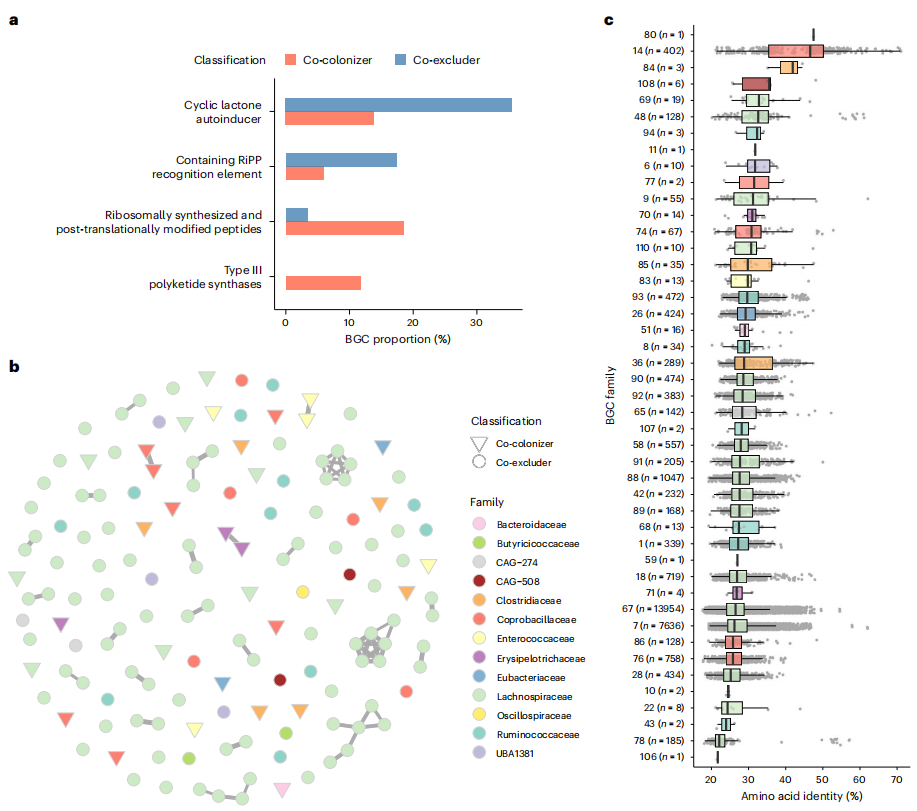
圖6. 共排斥物種擁有參與群體感應的生物合成基因簇
研究小結
本研究聚焦腸桿菌科物種與腸道微生物組其他成員間的關系,綜合采用多種研究方法,取得一系列重要發現。通過構建基因組規模代謝模型,深入分析代謝競爭和互補,揭示了生境篩選在腸道微生物定植和群落組裝中的關鍵作用。進一步的代謝分析表明,共定植物種與共排斥物種在代謝功能上存在顯著差異,這可能反映它們對腸道不同生態位的適應性差異。對生物合成基因簇的研究顯示,共排斥物種中與群體感應相關的基因簇顯著富集,這些基因簇或許在調控腸桿菌科的腸道定植和豐度方面發揮著重要作用。本研究為腸道微生物組的生態機制提供了新見解,有望為干預腸道微生物組以防治相關疾病提供理論支撐。
END
Peng 撰文
Winly 校稿