
期刊:Cell Death & Differentiation
影響因子:15.4
發表時間:2026年4月
通訊作者:南京醫科大學附屬南京第一醫院杜前明研究員、成都中醫藥大學附屬中西醫結合醫院張旭教授、南京醫科大學護理學院刁紅婷教授及南京第一醫院劉超教授
伯豪技術服務:單細胞RNA測序(scRNA-seq)
導語
結直腸癌(CRC)是全球第三大常見惡性腫瘤,免疫檢查點抑制劑雖改變了實體瘤治療格局,但僅約5%的CRC患者從中獲益,腫瘤微環境(TME)介導的免疫抑制是導致治療耐藥的核心瓶頸。腫瘤相關成纖維細胞(CAFs)作為TME中最為豐富的基質細胞,已被證實可重塑免疫細胞環境、抑制效應細胞功能。然而,CAFs的高度異質性使其在免疫逃逸中的具體功能亞群和分子機制長期未明。2026年3月,南京醫科大學作者在Cell Death & Differentiation發表研究成果,通過單細胞RNA測序首次鑒定出8種活化CAFs亞群,鎖定特異性表達PCSK6的CAFs功能亞群,系統闡明了CAFs來源的PCSK6通過與CRC細胞表面ACVR1B受體結合,激活p300乙酰轉移酶對PD-L1進行K263位點乙酰化修飾,進而通過Rab8/EHBP1L1膜轉運復合物驅動PD-L1在細胞膜的重分布,最終導致CD8+T細胞耗竭和免疫逃逸的完整分子機制。研究同時證實,ACVR1B拮抗劑TP0427736可有效阻斷該信號軸,恢復CD8+T細胞殺傷活性,為CRC免疫治療提供了新靶點與機制依據。
技術服務
單細胞RNA測序(scRNA-seq)
(技術服務由伯豪生物提供)
主要研究
1. scRNA-seq揭示CRC腫瘤微環境中CAFs異質性,鑒定PCSK6+ CAF功能亞群
腫瘤微環境的高度異質性是導致CRC治療失敗的核心原因之一,而CAFs作為TME中最豐富的基質成分,其表型和功能的多樣性長期被傳統Bulk RNA-seq所掩蓋。
作者對CRC臨床樣本進行單細胞RNA測序,基于無監督聚類分析,在腫瘤組織中鑒定出8種活化狀態的CAFs亞群。進一步分析發現,其中一個亞群特異性高表達前蛋白轉化酶枯草溶菌素6(PCSK6),約32.08%的CAFs呈現PCSK6陽性特征。與正常成纖維細胞相比,PCSK6在CRC腫瘤基質中顯著上調。免疫組化分析進一步驗證了這一發現:PCSK6蛋白在CRC腫瘤組織中的表達水平遠高于癌旁正常組織,且其表達水平與患者不良預后顯著相關。這一結果首次從單細胞精度揭示了CAFs的功能異質性,并錨定了PCSK6+ CAFs作為CRC免疫微環境重塑的關鍵功能亞群。
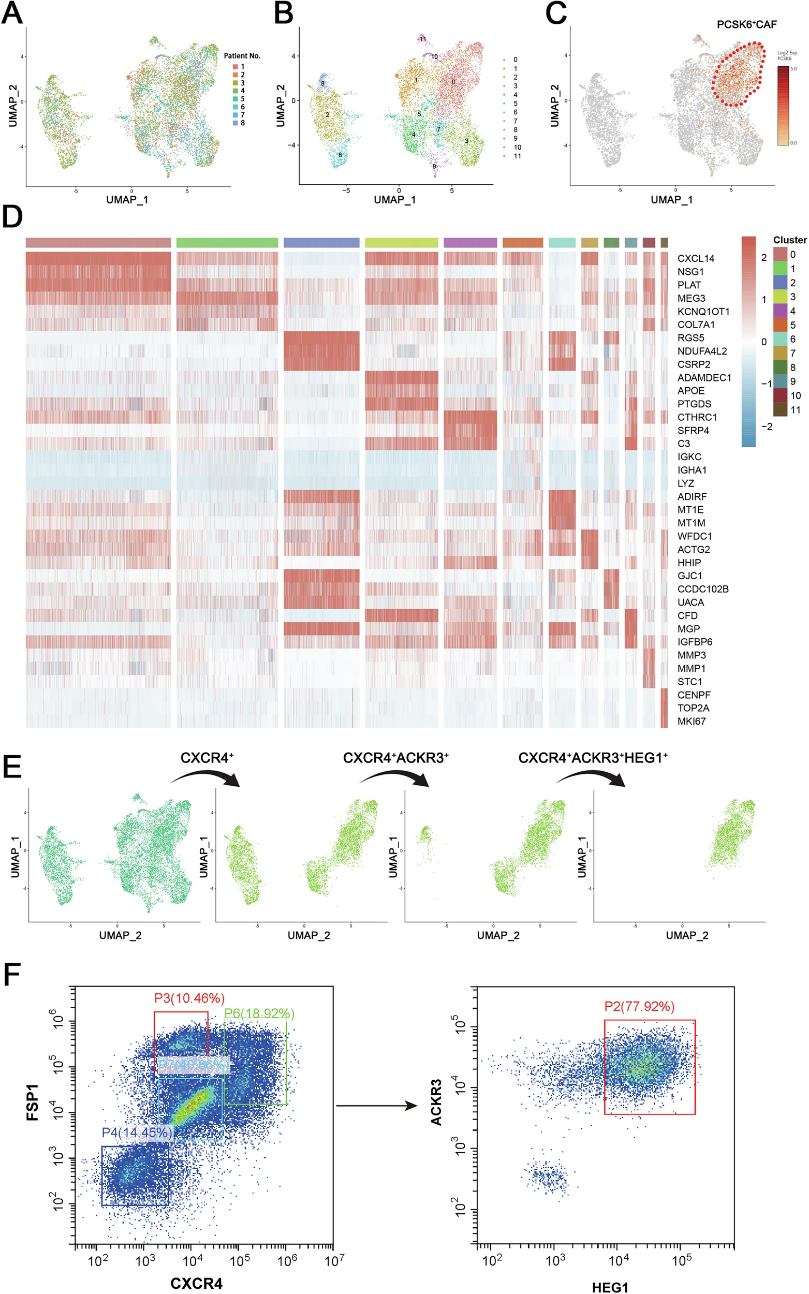
圖1 單細胞RNA測序揭示CRC中CAFs異質性
2. PCSK6+ CAFs通過誘導CD8+ T細胞耗竭促進CRC進展
為明確PCSK6+ CAFs在CRC進展中的功能角色,作者設計了系統的體內外功能驗證實驗。將PCSK6+ CAFs與CRC細胞共培養后發現,PCSK6+ CAFs能夠顯著增強CRC細胞的增殖與遷移能力;在皮下移植瘤模型中,PCSK6+ CAFs的引入導致腫瘤體積和重量均顯著增加,Ki-67染色證實腫瘤增殖活性增強。
免疫微環境分析顯示,PCSK6+ CAFs處理的腫瘤組織中,CD8+ T細胞的浸潤數量和活化標志物(Granzyme B、IFN-γ)表達水平均顯著下降,T細胞呈現出典型的耗竭表型(PD-1+TIM-3+雙陽性比例升高)。相反,當在CAFs中敲低PCSK6后,腫瘤生長受到明顯抑制,瘤內CD8+ T細胞的浸潤密度和殺傷活性均顯著恢復。這一正反實驗設計嚴謹地證實:PCSK6+ CAFs通過驅動CD8+ T細胞耗竭,發揮促腫瘤免疫功能。
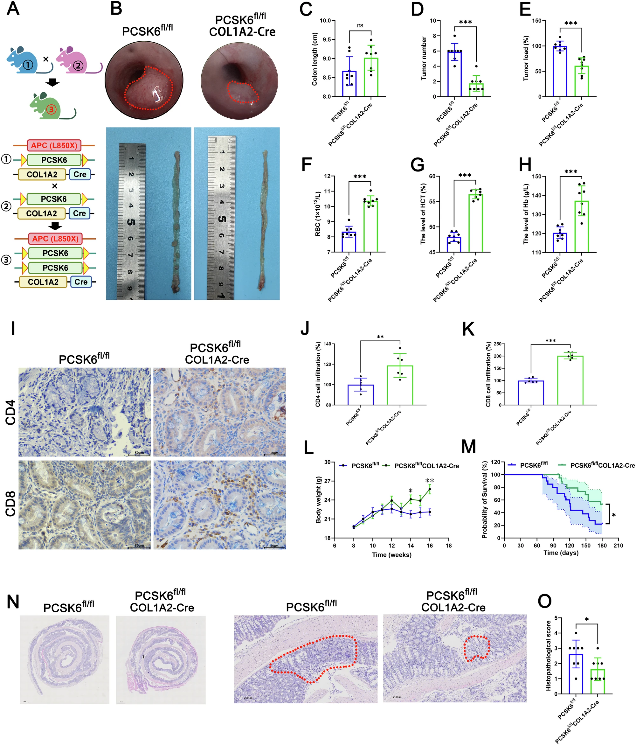
圖2 PCSK6+ CAFs促進CRC細胞增殖遷移,抑制CD8+ T細胞浸潤與活化
3. PCSK6與CRC細胞表面ACVR1B受體直接互作,啟動下游信號級聯
PCSK6作為一種分泌型前蛋白轉化酶,如何將信號從CAFs傳遞至CRC細胞?作者通過免疫共沉淀(Co-IP)和GST pull-down實驗鑒定出關鍵受體——激活素A受體1B型(ACVR1B)。實驗證實,CAFs分泌的PCSK6可直接與CRC細胞膜表面的ACVR1B受體相結合,這種配體-受體互作是啟動胞內信號級聯的關鍵分子事件。
為驗證ACVR1B的功能必要性,研究者利用CRISPR-Cas9技術敲除CRC細胞中的ACVR1B,發現PCSK6刺激所誘導的下游效應幾乎完全消失。進一步,使用ACVR1B特異性拮抗劑TP0427736處理后,PCSK6對PD-L1膜分布和CD8+ T細胞功能的調控作用被顯著逆轉。該結果清晰表明:PCSK6/ACVR1B信號軸是連接CAFs與CRC細胞免疫逃逸的核心通路。
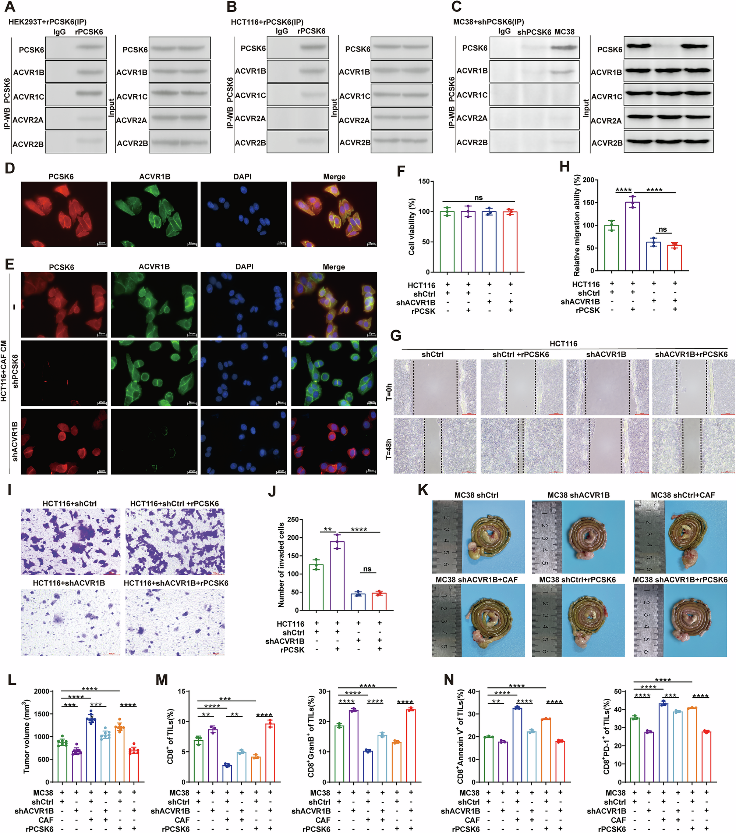
圖3 PCSK6直接結合CRC細胞表面ACVR1B受體,啟動促免疫逃逸信號級聯
4. p300乙酰轉移酶介導PD-L1 K263位點乙酰化,增強PD-L1膜分布
PD-L1作為關鍵的免疫檢查點分子,其功能不僅取決于表達量,更依賴于其在細胞膜上的定位分布。作者發現,PCSK6/ACVR1B信號激活后,可募集乙酰轉移酶p300至PD-L1分子,對其第263位賴氨酸殘基(K263)進行乙酰化修飾。體內外乙酰化實驗證實,PCSK6刺激可顯著增強PD-L1的乙酰化水平,而p300敲低則完全阻斷該效應。
值得注意的是,K263位點的乙酰化并不改變PD-L1的總蛋白表達量,而是通過構象變化影響其亞細胞定位。將K263突變為精氨酸(K263R,模擬去乙酰化狀態)后,PD-L1在細胞膜上的分布顯著減少,更多滯留于胞質中,證實K263乙酰化是決定PD-L1膜定位的關鍵翻譯后修飾。這一發現揭示了PD-L1活性調控的新維度——翻譯后修飾驅動的空間重分布機制,突破了傳統「表達量決定功能」的認知框架。
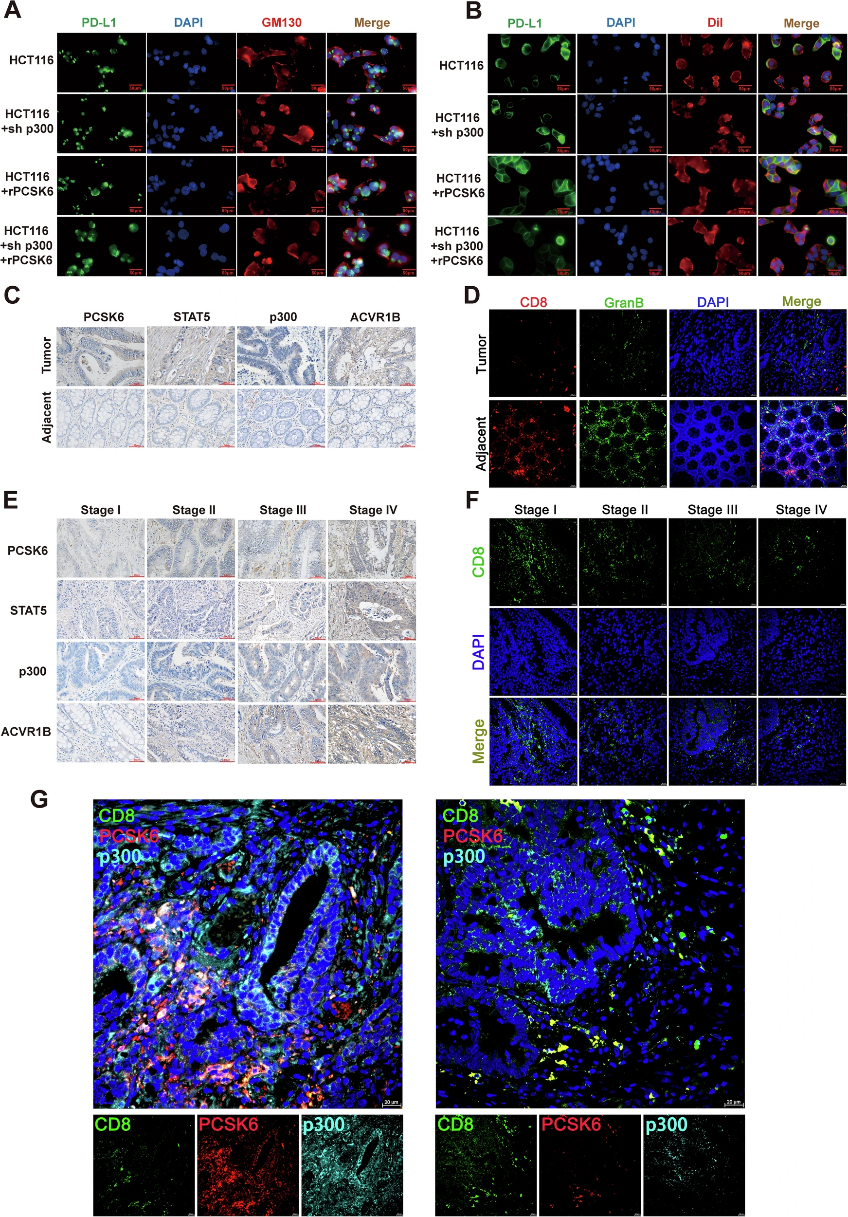
圖4 p300乙酰轉移酶催化PD-L1 K263位點乙酰化修飾,增強其細胞膜定位
5. Rab8/EHBP1L1膜轉運復合物驅動乙酰化PD-L1在細胞膜的重分布
乙酰化修飾后的PD-L1如何從胞質轉運至細胞膜?作者通過篩選發現,小GTP酶Rab8及其效應蛋白EHBP1L1構成的膜轉運復合物是介導這一過程的關鍵執行者。免疫共沉淀和免疫熒光共定位實驗顯示,乙酰化的PD-L1與Rab8/EHBP1L1復合物存在直接相互作用,且這一結合依賴于K263位點的乙酰化狀態。
抗體喂養實驗(antibody feeding assay)動態追蹤了PD-L1的內吞與再循環過程。結果顯示,在PCSK6刺激下,PD-L1的膜再循環速率顯著加快,細胞表面PD-L1的半衰期延長。Rab8敲低后,乙酰化PD-L1在胞質中異常積聚,膜定位信號大幅減弱。此外,研究者利用高爾基體標志物GM130進行共定位分析,發現PD-L1在Rab8/EHBP1L1復合物的護送下,經高爾基體-囊泡運輸途徑被精準遞送至細胞膜。該結果首次完整描繪了CAFs來源的PCSK6經「乙酰化-膜轉運」級聯實現PD-L1膜重分布的精細過程。
6. 靶向PCSK6/ACVR1B信號軸恢復抗腫瘤免疫——臨床轉化意義與治療策略
基于上述機制發現,作者進一步評估了該信號軸的臨床相關性。多中心CRC臨床隊列分析顯示,腫瘤組織中PCSK6高表達與CD8+ T細胞浸潤減少及患者總生存期縮短顯著相關;相反,較高的PD-L1膜表達水平與更差的免疫治療應答相關。值得注意的是,PCSK6表達與CD8+ T細胞浸潤呈負相關,而與PD-L1膜水平呈正相關,三者形成了具有預后指示價值的關聯網絡。
在治療層面,研究者在CRC荷瘤小鼠模型中驗證了ACVR1B拮抗劑TP0427736的治療潛力。TP0427736處理可顯著抑制PCSK6/ACVR1B-p300-Rab8/EHBP1L1-PD-L1信號軸的激活,降低PD-L1膜分布,促進瘤內CD8+ T細胞的浸潤和活化,最終顯著延緩腫瘤生長并延長小鼠生存期。聯合PD-1抗體治療后,抗腫瘤效應進一步增強,提示靶向該信號軸可與現有免疫檢查點抑制劑產生協同效應。該研究首次提出「CAFs來源的PCSK6作為上游免疫抑制信號啟動子」的概念,為CRC患者提供了基于腫瘤基質微環境的精準干預新策略。
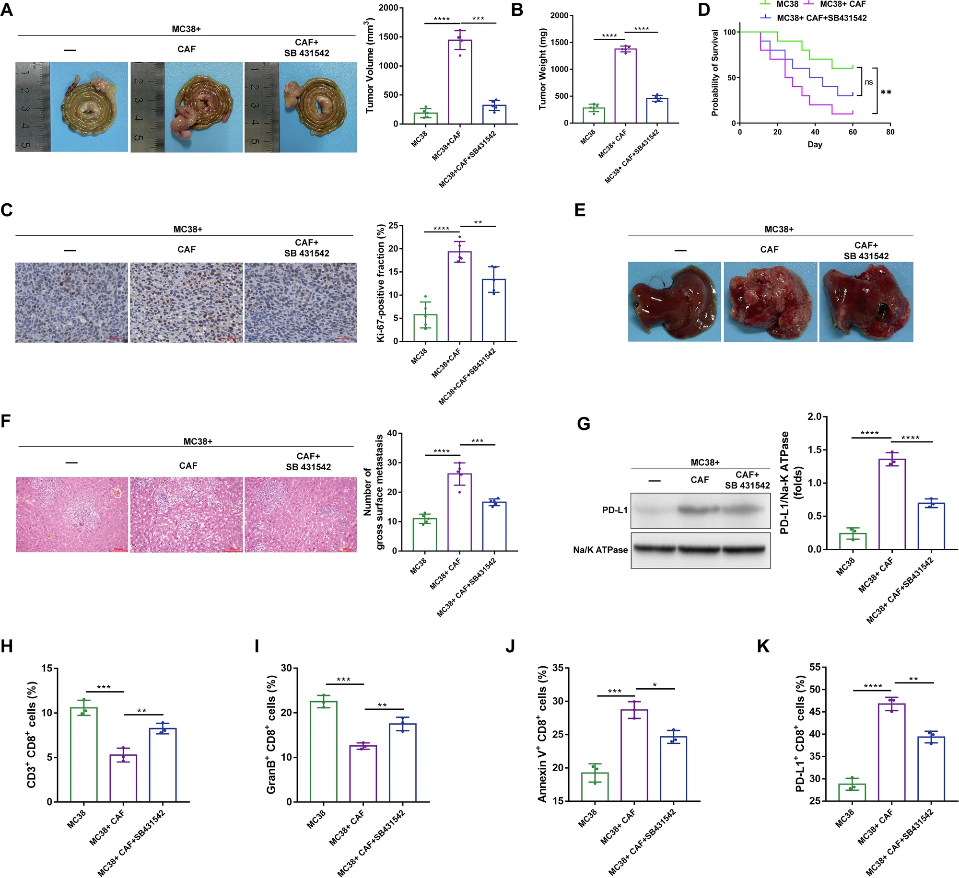
圖5 acvr1b特異性抑制劑SB431542在體內抑制免疫抑制和肝轉移
參考文獻:
Du Q, Fei Y, Li T, Wang D, Hu S, Yang Y, et al. (2026) Targeting CAFs-derived PCSK6 inhibits redistribution of PD-L1 and restores response of CD8+ T cells against colorectal cancer. Cell Death & Differentiation. https://doi.org/10.1038/s41418-026-01736-3