
英文標(biāo)題:Metabolic signaling of ceramides through the FPR2 receptor inhibits adipocyte thermogenesis
中文標(biāo)題:神經(jīng)酰胺通過FPR2介導(dǎo)的代謝信號(hào)傳導(dǎo)抑制脂肪細(xì)胞產(chǎn)熱
發(fā)表期刊:Science
影響因子:44.7
研究背景
神經(jīng)酰胺是鞘磷脂代謝關(guān)鍵產(chǎn)物,與多種代謝疾病密切相關(guān),但其作為系統(tǒng)信號(hào)分子的體內(nèi)作用機(jī)制尚不明晰。脂肪細(xì)胞產(chǎn)熱對(duì)維持機(jī)體能量平衡和體溫穩(wěn)態(tài)至關(guān)重要,cAMP信號(hào)通路參與其中,G蛋白偶聯(lián)受體(GPCRs)在調(diào)控脂肪細(xì)胞產(chǎn)熱中起核心作用。目前,雖已深入研究GPCRs對(duì)脂肪細(xì)胞產(chǎn)熱的調(diào)控,但神經(jīng)酰胺與GPCRs的交互關(guān)系及對(duì)脂肪細(xì)胞產(chǎn)熱的調(diào)節(jié)作用研究較少。本研究聚焦于此,期望揭示相關(guān)調(diào)控機(jī)制,為解析神經(jīng)酰胺在代謝疾病中的作用機(jī)制提供新思路,助力開發(fā)代謝疾病創(chuàng)新治療策略。
研究成果
1、C16:0神經(jīng)酰胺對(duì)脂肪代謝相關(guān)功能及FPR2受體信號(hào)通路的影響
研究者以C16:0神經(jīng)酰胺為例,研究神經(jīng)酰胺是否直接與GPCRs相互作用,啟動(dòng)跨膜信號(hào)傳導(dǎo),調(diào)節(jié)棕色和米色脂肪細(xì)胞的產(chǎn)熱功能(圖1A)。給正常飲食(NCD)喂養(yǎng)的野生型(WT)小鼠腹腔注射溶劑或C16:0神經(jīng)酰胺,持續(xù)3天。在冷暴露期間,給予外源性C16:0神經(jīng)酰胺會(huì)降低小鼠的耗氧量和能量消耗,但不影響其食物攝入量和運(yùn)動(dòng)活性(圖1B-C)。
細(xì)胞實(shí)驗(yàn)方面,研究者使用C16:0神經(jīng)酰胺對(duì)棕色脂肪組織(BAT)、皮下白色脂肪組織(scWAT)以及成熟脂肪細(xì)胞進(jìn)行短暫刺激(刺激時(shí)長(zhǎng)為15分鐘)時(shí),這些組織和細(xì)胞內(nèi)的cAMP含量呈現(xiàn)出明顯的下降趨勢(shì)(圖1D),這提示Gi偶聯(lián)受體可能參與了神經(jīng)酰胺對(duì)脂肪組織的急性作用。
為進(jìn)一步探究,研究者借助GSE40486的微陣列數(shù)據(jù),篩選出小鼠棕色脂肪組織中高表達(dá)的60種GPCRs。后續(xù)利用GloSensor-cAMP檢測(cè)法對(duì)這些受體展開篩選,發(fā)現(xiàn)在過表達(dá)FPR2的人胚腎293(HEK293)細(xì)胞中加入C16:0神經(jīng)酰胺后,由佛司可林誘導(dǎo)產(chǎn)生的cAMP水平呈劑量依賴性下降(圖1E)。使用Gαi1和Gαi2解離實(shí)驗(yàn)進(jìn)一步驗(yàn)證,結(jié)果表明人源hFPR2和小鼠源mFPR2這兩種不同物種的GPCRs對(duì)其內(nèi)源配體的反應(yīng)有時(shí)會(huì)有所不同(圖1F)。具體而言,C16:0神經(jīng)酰胺在Gi信號(hào)通路中激活hFPR2或mFPR2的效能與其他已知的內(nèi)源性配體(如fMLFII、Ac2-26、LL-37、humanin)相當(dāng),但功效較低,可能是FPR2的部分激動(dòng)劑。
研究者運(yùn)用G蛋白解離和β-arrestin1募集實(shí)驗(yàn),探索原代脂肪細(xì)胞內(nèi)源性FPR2的響應(yīng)。構(gòu)建含G蛋白及β-arrestin1 BRET探針的慢病毒感染原代脂肪細(xì)胞,并以Adipoq-Cre+/−Fpr2fl/fl小鼠作陰性對(duì)照。結(jié)果顯示,從Fpr2fl/fl小鼠分離的原代脂肪細(xì)胞,其Gi信號(hào)通路可被激活,且對(duì)C16:0神經(jīng)酰胺、LL-37和fMLFII呈濃度依賴性反應(yīng),而陰性對(duì)照的原代脂肪細(xì)胞無此反應(yīng)(圖1G)。
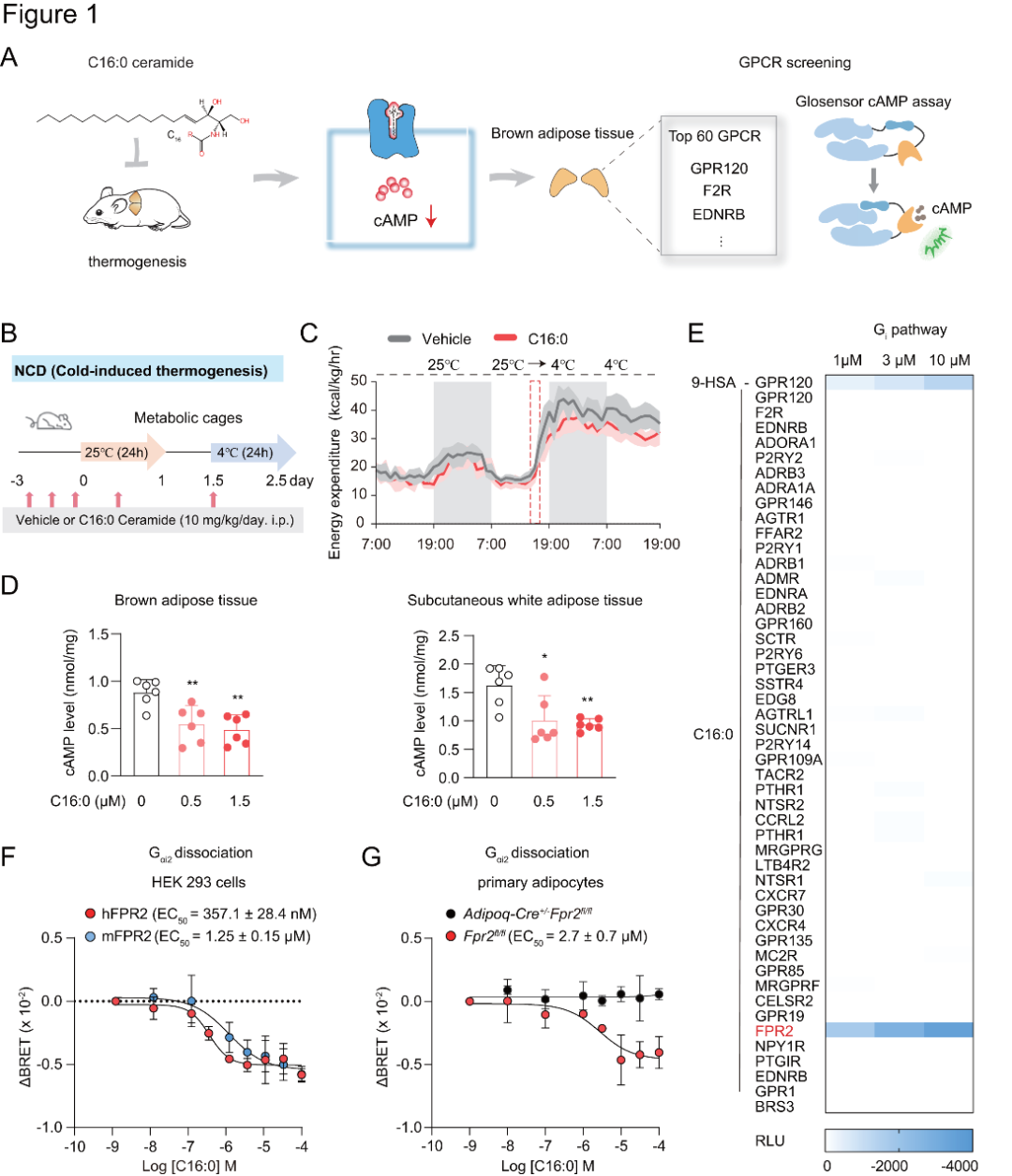
圖1.神經(jīng)酰胺膜受體的鑒定
2、FPR2與神經(jīng)酰胺的直接相互作用
研究者使用FPR2抗體,研究了野生型小鼠的scWAT和BAT中FPR2的表達(dá)模式,并以Fpr2基因缺陷型(Sox2-CreER+/−Fpr2fl/fl)小鼠作為陰性對(duì)照。結(jié)果表明,F(xiàn)PR2主要分布在野生型小鼠的scWAT或BAT中表達(dá)脂聯(lián)素的脂肪細(xì)胞內(nèi),而在Fpr2基因缺陷型小鼠中則不存在這種分布(圖2A-B)。
為探究C16:0神經(jīng)酰胺與FPR2的直接結(jié)合特性,研究者采用兩種方法,(1)體外實(shí)驗(yàn):在競(jìng)爭(zhēng)性結(jié)合實(shí)驗(yàn)中,用FITC標(biāo)記的高親和力肽激動(dòng)劑WK(FITC)YMVm作用于FPR2。構(gòu)建含Nanoluc熒光基序且與hFPR2的N端融合的質(zhì)粒,在HEK293細(xì)胞中表達(dá),檢測(cè)WK(FITC)YMVm的結(jié)合情況。發(fā)現(xiàn)C16:0神經(jīng)酰胺濃度增加時(shí),WK(FITC)YMVm與Nluc-FPR2間的BRET信號(hào)減弱;(2)體內(nèi)實(shí)驗(yàn):合成125I標(biāo)記的FPR2肽激動(dòng)劑125I-Ac2-26。飽和結(jié)合分析顯示,其能特異性結(jié)合野生型小鼠BAT、scWAT及過表達(dá)hFPR2的HEK293細(xì)胞的膜組分。競(jìng)爭(zhēng)性結(jié)合實(shí)驗(yàn)證實(shí),C16:0神經(jīng)酰胺可與125I-Ac2-26競(jìng)爭(zhēng)結(jié)合上述膜組分(圖2C)。
已知GPCR結(jié)合會(huì)使細(xì)胞外結(jié)構(gòu)域發(fā)生選擇性構(gòu)象變化,為探究FPR2是否如此,研究者構(gòu)建FlAsH BRET傳感器,監(jiān)測(cè)其細(xì)胞外環(huán)(ECL)區(qū)域在結(jié)合神經(jīng)酰胺和fMLFII時(shí)的構(gòu)象變化(圖2D)。結(jié)果顯示,C16:0神經(jīng)酰胺使FPR2的N端靠近ECL1和ECL2區(qū)域,而fMLFII則使FPR2的N端遠(yuǎn)離ECL2區(qū)域(圖2E)。這表明C16:0神經(jīng)酰胺與FPR2結(jié)合會(huì)引發(fā)受體特定細(xì)胞外構(gòu)象變化,可能影響其信號(hào)傳導(dǎo)特異性。
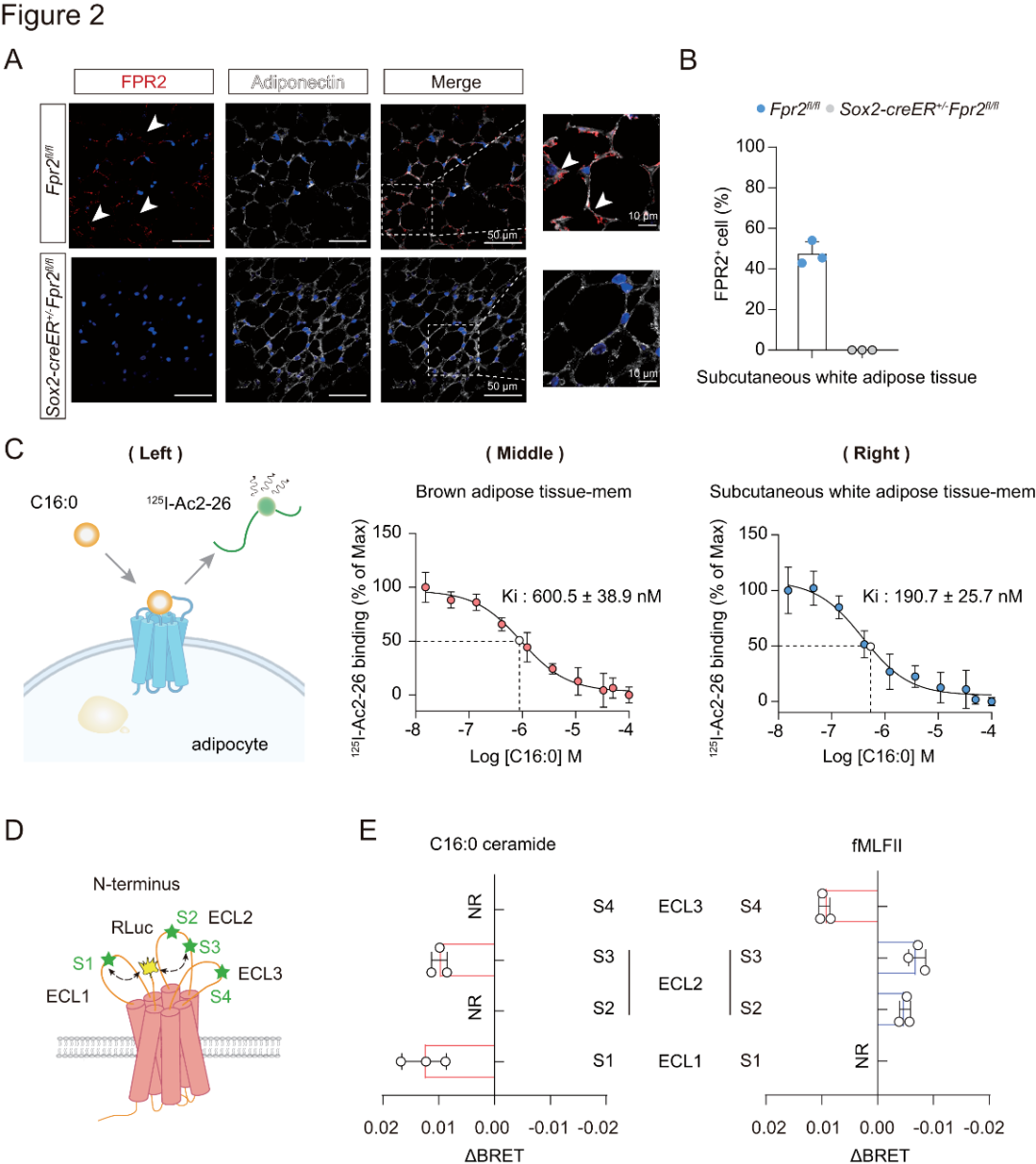
圖2.脂肪細(xì)胞中C16:0神經(jīng)酰胺與FPR2的直接相互作用
3、FPR2介導(dǎo)C16:0神經(jīng)酰胺對(duì)產(chǎn)熱的抑制作用
為探究C16:0神經(jīng)酰胺對(duì)冷誘導(dǎo)產(chǎn)熱的抑制是否依賴脂肪細(xì)胞FPR2表達(dá),研究者構(gòu)建了Ucp1-Cre+/-Fpr2fl/fl小鼠及Adipoq-Cre+/-Fpr2fl/fl小鼠。對(duì)兩類基因編輯小鼠及對(duì)照Fpr2fl/fl小鼠,喂正常飲食,腹腔注射溶劑或C16:0神經(jīng)酰胺(圖3A)。結(jié)果顯示,C16:0神經(jīng)酰胺可抑制Fpr2fl/fl小鼠在室溫和寒冷下的能量消耗與耗氧量,但在Adipoq-Cre+/-Fpr2fl/fl及Ucp1-Cre+/-Fpr2fl/fl小鼠中無此效果(圖3B-C)。
研究者進(jìn)一步研究了冷暴露后C16:0神經(jīng)酰胺通過FPR2對(duì)脂肪細(xì)胞產(chǎn)熱的影響。給喂食正常飲食的Adipoq-Cre+/-Fpr2fl/fl小鼠、Ucp1-Cre+/-Fpr2fl/fl小鼠以及對(duì)照Fpr2fl/fl小鼠腹腔注射溶劑或C16:0神經(jīng)酰胺。為探究C16:0神經(jīng)酰胺對(duì)BAT活性和米色脂肪生成的抑制作用是否依賴于FPR2,在冷暴露一天后檢測(cè)了小鼠BAT和scWAT中產(chǎn)熱相關(guān)基因的相對(duì)表達(dá)(圖3D)。C16:0神經(jīng)酰胺處理降低了對(duì)照組小鼠的直腸溫度以及腹股溝和肩胛間區(qū)域的局部溫度,同時(shí)降低了UCP-1蛋白水平和產(chǎn)熱基因的表達(dá)(圖3E-F)。H&E染色和免疫組化進(jìn)一步證實(shí),C16:0神經(jīng)酰胺處理抑制了Fpr2fl/fl小鼠的BAT活性和米色脂肪生成(圖3G)。然而,在兩類基因編輯小鼠中,C16:0神經(jīng)酰胺的上述作用消失了(圖3E-G)。白色脂肪組織中Fpr2基因缺乏可上調(diào)UCP-1等產(chǎn)熱相關(guān)基因表達(dá),且不依賴外源性C16:0神經(jīng)酰胺(圖3D-G)。綜上,在生理?xiàng)l件下,內(nèi)源性神經(jīng)酰胺可能通過激活FPR2抑制白色脂肪組織向棕色脂肪的轉(zhuǎn)化及產(chǎn)熱過程(圖3呈現(xiàn)了Adipoq-Cre+/-Fpr2fl/fl小鼠在相關(guān)實(shí)驗(yàn)中的結(jié)果,Ucp1-Cre+/-Fpr2fl/fl小鼠的實(shí)驗(yàn)結(jié)果詳情可查閱原文的補(bǔ)充材料)。

圖3.C16:0神經(jīng)酰胺通過FPR2抑制冷暴露誘導(dǎo)的產(chǎn)熱作用
4、FPR2與神經(jīng)酰胺復(fù)合物的整體結(jié)構(gòu)及識(shí)別特異性
神經(jīng)酰胺的脂肪鏈長(zhǎng)度以及特定位置上不飽和的碳碳雙鍵存在差異,會(huì)影響其生理功能。因此,研究者探究了各種脂質(zhì)結(jié)構(gòu)對(duì)FPR2的作用活性(圖4A-B)。神經(jīng)酰胺的亞結(jié)構(gòu),如棕櫚酸(PA)、花生酸(AA)和鞘氨醇,不能激活FPR2下游的Gi信號(hào)通路(圖4B)。只有具有飽和脂肪鏈(C2:0、C6:0、C10:0、C16:0、C18:0和C20:0)的神經(jīng)酰胺才能激活FPR2(圖4B)。帶有不飽和碳碳雙鍵(C18:1和C24:1)的神經(jīng)酰胺以及超長(zhǎng)鏈神經(jīng)酰胺無法激活FPR2(圖4B)。
為了剖析飽和長(zhǎng)鏈神經(jīng)酰胺是如何被膜受體FPR2特異性識(shí)別的,研究者使用單顆粒冷凍電子顯微鏡(cryo-EM)來確定與神經(jīng)酰胺和Gi1形成復(fù)合物的人類FPR2的結(jié)構(gòu),重構(gòu)、純化了C16:0、C18:0和C20:0神經(jīng)酰胺-FPR2-Gi1-scFv16復(fù)合物(圖4C)。神經(jīng)酰胺呈 “p” 形垂直插入FPR2正構(gòu)結(jié)合位點(diǎn),與fMLFII和humanin結(jié)合方式相似,區(qū)別于與Gq偶聯(lián)的神經(jīng)酰胺受體CYSLTR2(圖4D)。
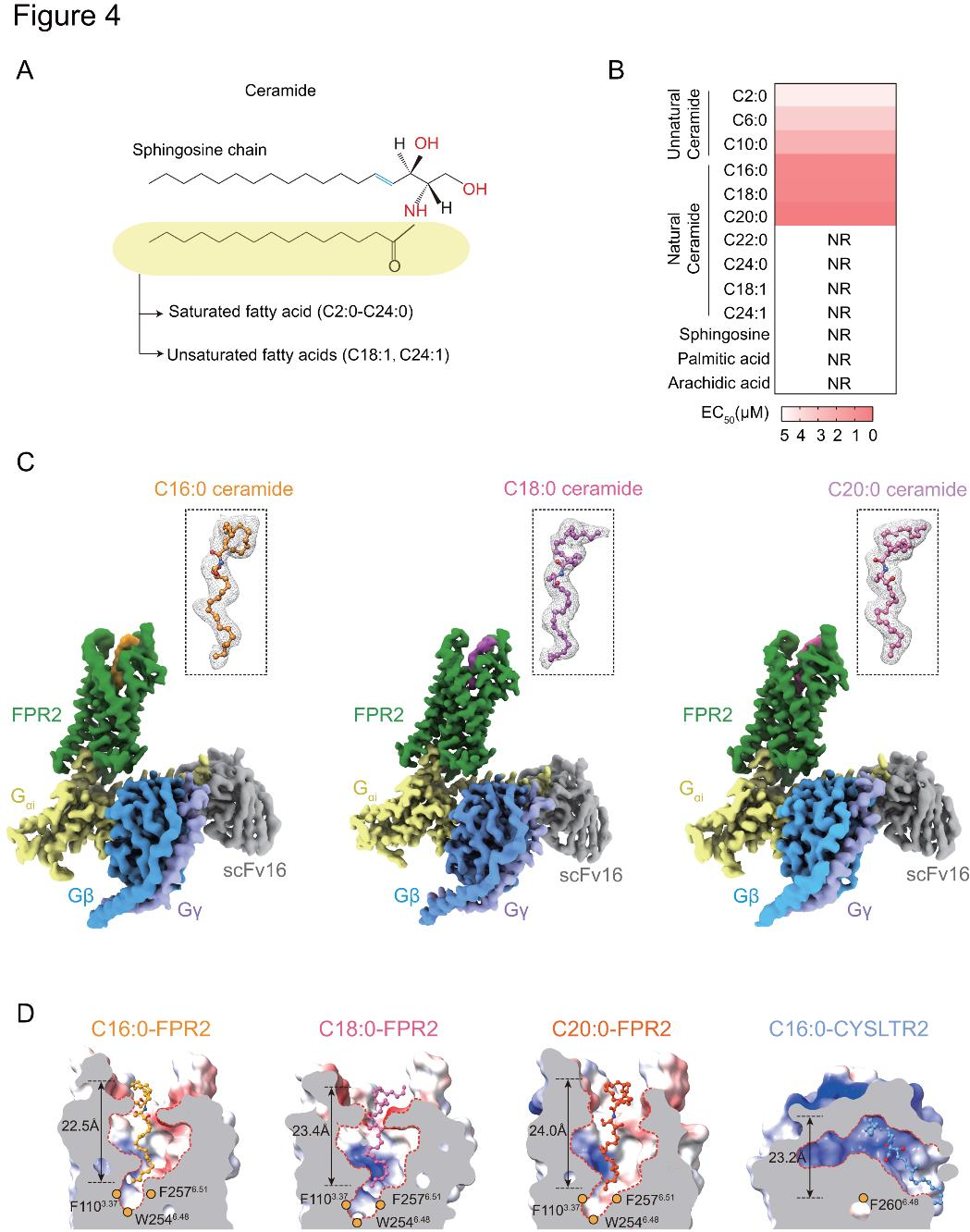
圖4.hFPR2-Gi配合物與神經(jīng)酰胺結(jié)合的整體結(jié)構(gòu)
5、神經(jīng)酰胺在FPR2正構(gòu)位點(diǎn)的結(jié)合
在C16:0、C18:0和C20:0神經(jīng)酰胺-FPR2-Gi1復(fù)合物結(jié)構(gòu)中,神經(jīng)酰胺由C18鞘氨醇鏈與N-酰基鏈經(jīng)中心酰胺鍵連接,其鞘氨醇脂肪鏈深入FPR2配體口袋,與特定芳香殘基作用并占據(jù)fMLFII位置(圖4D)。脂肪鏈位于由L109³.³?、F110³.³?、R201?.³?、R205?.?²和F257?.?¹組成的口袋中(圖5A-D)。L109³.³?A、F110³.³?A、R201?.³?A、R205?.?²A和F257?.?¹A的突變會(huì)降低神經(jīng)酰胺誘導(dǎo)的FPR2激活(圖5F)。特別是,H102³.²?和F178ECL2可能與神經(jīng)酰胺的不飽和C=C雙鍵形成長(zhǎng)距離的π-π堆積(圖5A-D)。H102³.²?L/A或 F178ECL2L/A的突變顯著降低了這些神經(jīng)酰胺誘導(dǎo)的FPR2激活(圖5F)。脂肪酸部分脂肪鏈折疊成特定結(jié)構(gòu),圍繞其疏水口袋殘基突變會(huì)損害FPR2對(duì)神經(jīng)酰胺刺激的激活,表明FPR2細(xì)胞外疏水環(huán)境有助于結(jié)合(圖5F)。由于FPR2的配體口袋較大,且神經(jīng)酰胺的脂肪鏈具有柔性,研究者進(jìn)行了分子動(dòng)力學(xué)模擬,以研究在冷凍電鏡結(jié)構(gòu)中未觀察到的潛在相互作用。發(fā)現(xiàn)E89ECL1等殘基與神經(jīng)酰胺中心羧化鞘氨醇基團(tuán)相互作用(圖5E)。這些殘基的丙氨酸突變降低了FPR2對(duì)神經(jīng)酰胺刺激的激活(圖5F)。
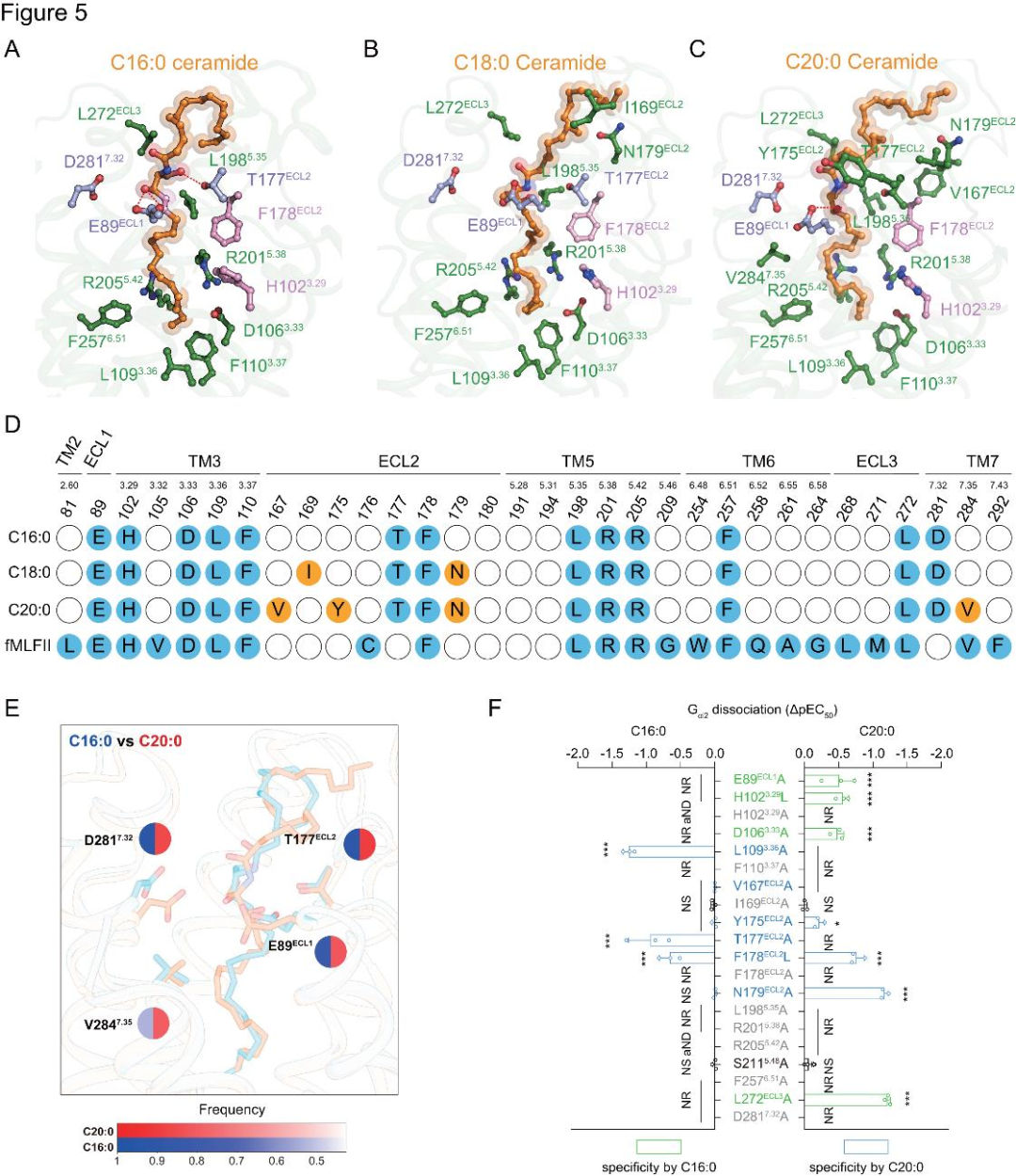
圖5.FPR2對(duì)神經(jīng)酰胺的識(shí)別
6、FPR2對(duì)神經(jīng)酰胺識(shí)別特異性的調(diào)控機(jī)制
在C16:0(p構(gòu)象)、C18:0(s構(gòu)象)及C20:0(s構(gòu)象)神經(jīng)酰胺與FPR2相關(guān)復(fù)合物結(jié)構(gòu)中,神經(jīng)酰胺脂肪酸部分呈彎曲構(gòu)型(圖4C和圖5A-C),而脂肪酸鏈中C=C雙鍵的存在可能會(huì)破壞此構(gòu)型,致不飽和神經(jīng)酰胺無法與FPR2穩(wěn)定互作(圖6C-D)。飽和Cn:0神經(jīng)酰胺活性隨脂肪酸碳鏈增長(zhǎng)而增強(qiáng),達(dá)22個(gè)碳原子時(shí)活性消失(圖4A-B)。在相關(guān)復(fù)合物中,神經(jīng)酰胺脂肪酸脂肪鏈被跨膜結(jié)構(gòu)域5(TM5)、ECL2和ECL3細(xì)胞外疏水殘基包圍(圖6A),碳鏈增長(zhǎng)可增加與FPR2疏水作用(圖6B)。這些殘基突變對(duì)C18:0、C20:0誘導(dǎo)的FPR2激活影響大于C16:0,表明FPR2區(qū)域疏水口袋可塑性對(duì)容納長(zhǎng)鏈神經(jīng)酰胺很重要,且計(jì)算模擬顯示該口袋對(duì)極長(zhǎng)鏈神經(jīng)酰胺而言可能過小(圖6E)。
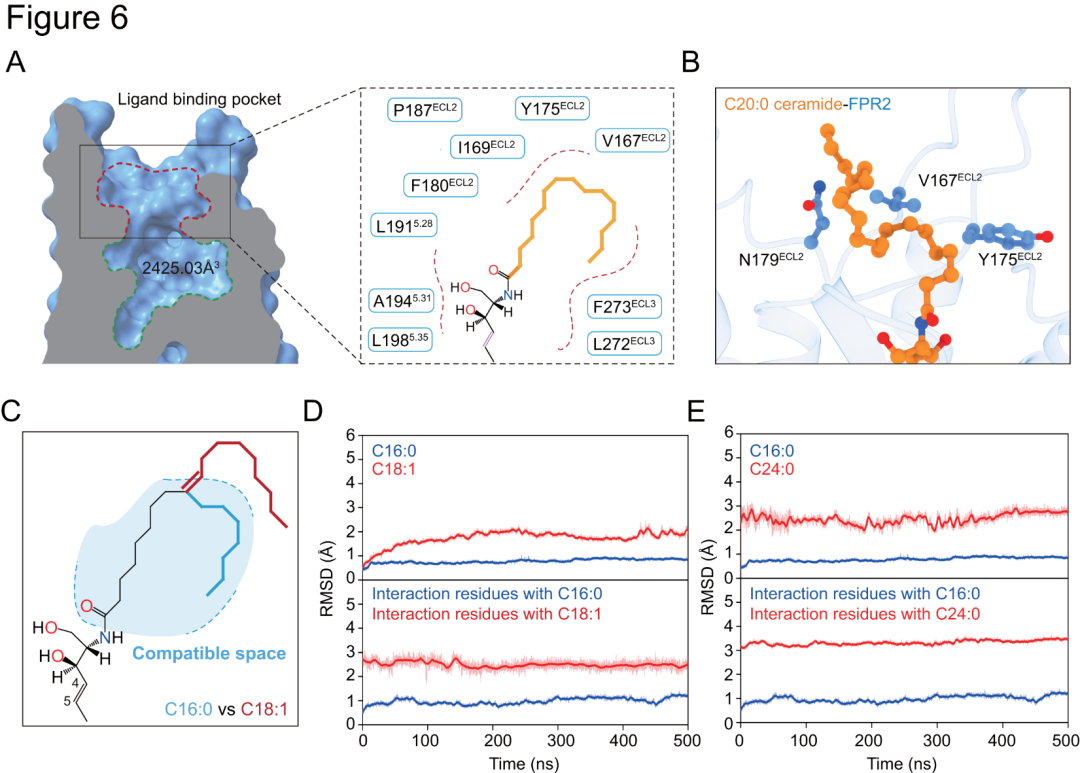
圖6.FPR2對(duì)不同神經(jīng)酰胺選擇性識(shí)別的結(jié)構(gòu)基礎(chǔ)
7、神經(jīng)酰胺對(duì)FPR2及其相關(guān)受體的選擇性作用
為探究神經(jīng)酰胺信號(hào)傳導(dǎo)特異性,研究者測(cè)試了C16:0神經(jīng)酰胺對(duì)6種與FPR2密切相關(guān)的GPCR(FPR3、FPR1、GPR32、C5R1、CMKLR1和GPR1)的活性影響。結(jié)果顯示,這些受體對(duì)C16:0神經(jīng)酰胺均無響應(yīng)(圖7A-B)。其中,F(xiàn)PR1和FPR3與FPR2親緣關(guān)系最近(圖7C),但FPR2中識(shí)別羧化鞘氨醇基團(tuán)或不飽和C=C的關(guān)鍵殘基,如E89ECL1、D281?.³²和H102³.²?,在FPR1中被替換(圖7C-D),相應(yīng)突變降低了神經(jīng)酰胺對(duì)FPR2的激活,致使神經(jīng)酰胺無法激活FPR1(圖7F)。FPR3中L198?.³?、R201?.³?和R205?.?²發(fā)生替換(圖7C和E),F(xiàn)PR2中對(duì)應(yīng)突變也降低C16:0神經(jīng)酰胺的激活作用(圖7F)。除FPR成員外,其他進(jìn)化上接近FPR2的受體無“EECL1&H³.²?”基序,F(xiàn)PR2相應(yīng)突變損害C16:0神經(jīng)酰胺誘導(dǎo)的Gi活性(圖7C和F)。相反,F(xiàn)PR1的G89ECL1E突變和 FPR3的A198?.³?L-H205?.?²R組合突變,使這兩種受體能夠感知C16:0神經(jīng)酰胺(圖7G-J)。以上結(jié)果確定E89ECL1、L198?.³?和R205?.?²為識(shí)別神經(jīng)酰胺的關(guān)鍵決定因素。
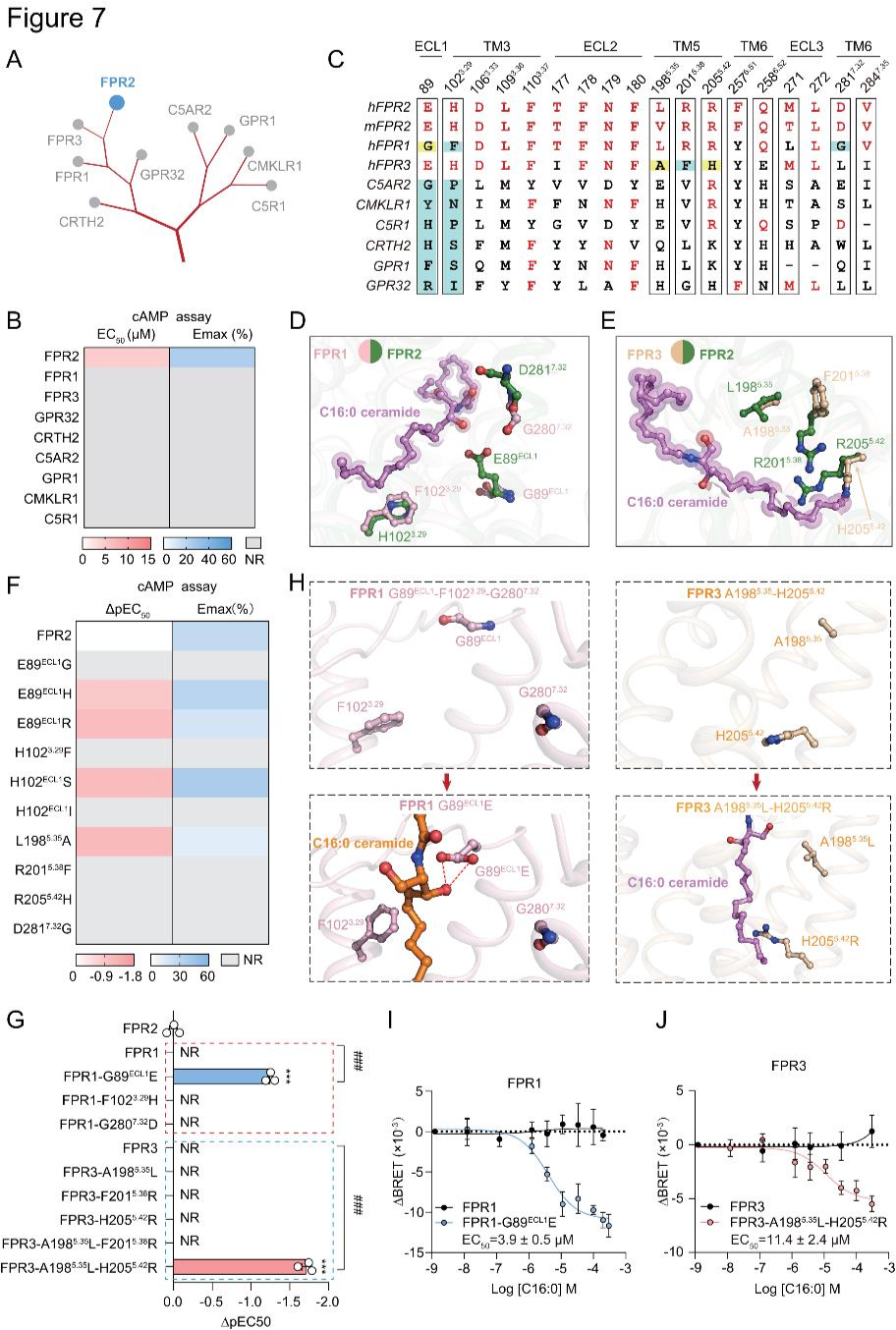
圖7.FPR2是C16:0神經(jīng)酰胺的特異性受體
8、神經(jīng)酰胺激活FPR2的潛在機(jī)制
為了探究神經(jīng)酰胺激活FPR2的機(jī)制,研究者基于AlphaFold蛋白質(zhì)結(jié)構(gòu)數(shù)據(jù)庫預(yù)測(cè)結(jié)構(gòu),在脂雙層中進(jìn)行分子動(dòng)力學(xué)松弛處理后,構(gòu)建了無配體FPR2模型(圖8A-B)。通過對(duì)比神經(jīng)酰胺-FPR2-Gi復(fù)合物的冷凍電鏡結(jié)構(gòu)與該模型,發(fā)現(xiàn)關(guān)鍵構(gòu)象變化(圖8C-G)。C16:0神經(jīng)酰胺的鞘氨醇脂肪鏈與特定殘基相互作用,引發(fā)殘基旋轉(zhuǎn)、移動(dòng),促使作為轉(zhuǎn)換開關(guān)的W254?.??與保守基序堆積,使神經(jīng)酰胺結(jié)合誘導(dǎo)的構(gòu)象變化經(jīng)轉(zhuǎn)換開關(guān)傳至相關(guān)基序及細(xì)胞質(zhì)區(qū)域(圖8E)。這一過程中,無配體FPR2結(jié)構(gòu)中的特定鹽橋和氫鍵被破壞,新的極性相互作用網(wǎng)絡(luò)形成(圖8F)。同時(shí),C16:0-FPR2-Gi結(jié)構(gòu)中,F(xiàn)PR2的TM7向外移動(dòng),引發(fā)相關(guān)基序重排,特定殘基間形成額外的π-π堆積和π-陽離子堆積(圖8G)。
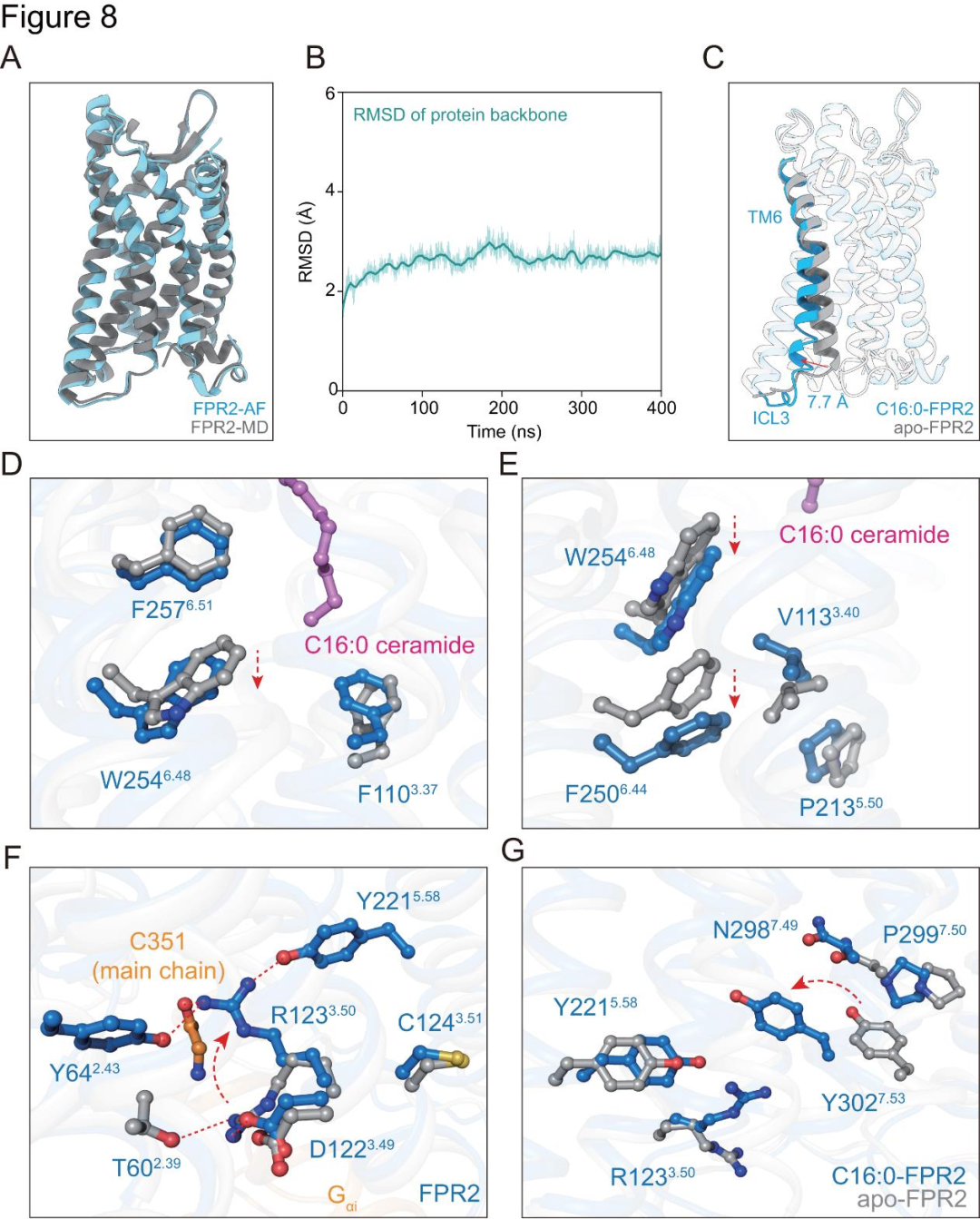
圖8.FPR2與C16:0神經(jīng)酰胺結(jié)合的激活機(jī)制
研究小結(jié)
神經(jīng)酰胺作為鞘磷脂代謝核心分子,其異常累積與糖尿病、肥胖及動(dòng)脈粥樣硬化等代謝疾病密切相關(guān)。近期研究發(fā)現(xiàn),C16:0神經(jīng)酰胺可以直接與脂肪細(xì)胞中的神經(jīng)酰胺膜受體FPR2結(jié)合,激活Gi信號(hào)通路,降低細(xì)胞內(nèi)cAMP水平,進(jìn)而抑制UCP-1表達(dá),降低產(chǎn)熱能力,基因敲除或拮抗劑實(shí)驗(yàn)已證實(shí)FPR2的介導(dǎo)作用。研究團(tuán)隊(duì)利用冷凍電鏡解析神經(jīng)酰胺-FPR2-Gi復(fù)合物結(jié)構(gòu),揭示FPR2對(duì)神經(jīng)酰胺的選擇性識(shí)別機(jī)制。該研究成果為深入剖析神經(jīng)酰胺的生理功能打開了全新的窗口,為代謝性疾病靶向治療開拓了新方向。
END
Peng 撰文
Orianna 校稿