前言
2025年10月10日,國務(wù)院正式公布《生物醫(yī)學(xué)新技術(shù)臨床研究和臨床轉(zhuǎn)化應(yīng)用管理?xiàng)l例》(國務(wù)院令第818號,下稱《條例》),并將于2026年5月1日起施行。
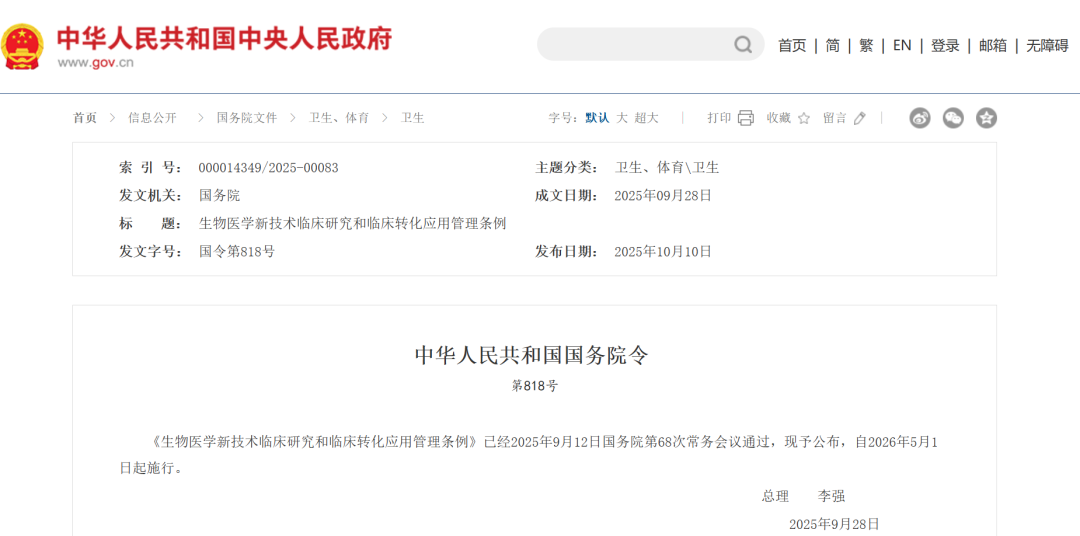
該條例是我國首次以國務(wù)院行政法規(guī)的形式,在國家層面構(gòu)建了覆蓋生物醫(yī)學(xué)新技術(shù)研發(fā)、臨床研究到臨床應(yīng)用全過程的法律體系。其出臺為蓬勃發(fā)展的生物醫(yī)學(xué)新技術(shù)領(lǐng)域提供了首部綜合性法規(guī)框架,在法規(guī)層級、管理范圍、審批機(jī)制、主體參與、法律責(zé)任等方面實(shí)現(xiàn)了多項(xiàng)突破。對于業(yè)界而言,它不僅是監(jiān)管制度的完善,更釋放出鼓勵創(chuàng)新與確保安全并重的強(qiáng)烈信號。
01 政策背景:創(chuàng)新熱潮與監(jiān)管挑戰(zhàn)
當(dāng)今全球生物醫(yī)學(xué)技術(shù)日新月異,新興技術(shù)在疾病預(yù)防、診斷、治療領(lǐng)域展現(xiàn)巨大潛力,也深刻改變著傳統(tǒng)醫(yī)療模式。中國在細(xì)胞與基因治療(CGT)、類器官等部分前沿領(lǐng)域已躋身全球科技前沿,許多突破性成果為疑難重癥和罕見病帶來新希望。然而,醫(yī)學(xué)創(chuàng)新往往快于監(jiān)管完善:一方面,新技術(shù)潛在倫理爭議和風(fēng)險容易引發(fā)社會質(zhì)疑;另一方面,不少科研成果難以真正惠及患者,新技術(shù)從實(shí)驗(yàn)室走向臨床的路徑不清晰。例如,此前震動業(yè)界的“基因編輯嬰兒”事件、各地未經(jīng)批準(zhǔn)的干細(xì)胞療法收費(fèi)應(yīng)用等,都暴露出監(jiān)管空白和風(fēng)險隱患。
在此背景下,國家高度重視生物醫(yī)學(xué)技術(shù)創(chuàng)新發(fā)展,過去數(shù)年陸續(xù)探索監(jiān)管機(jī)制:2015年原衛(wèi)計(jì)委和食藥監(jiān)總局發(fā)布《干細(xì)胞臨床研究管理辦法(試行)》,以醫(yī)療機(jī)構(gòu)為主體建立了干細(xì)胞臨床研究的規(guī)范路徑;海南博鰲樂城先行區(qū)也試點(diǎn)了細(xì)胞基因療法的轉(zhuǎn)化應(yīng)用特別監(jiān)管模式(少量數(shù)據(jù)+專家評審+患者知情同意)。但總體而言,生物醫(yī)學(xué)新技術(shù)領(lǐng)域一直缺乏上位法條例。《條例》的制定正是順應(yīng)時代發(fā)展、回應(yīng)人民關(guān)切之產(chǎn)物,填補(bǔ)了該領(lǐng)域法規(guī)空白,標(biāo)志著我國生物醫(yī)學(xué)新技術(shù)發(fā)展進(jìn)入法治化、規(guī)范化新軌道。

2025年3月10日,博鰲樂城公布第二批17項(xiàng)細(xì)胞與基因治療技術(shù)轉(zhuǎn)化應(yīng)用實(shí)施目錄
02 核心定義:首次明確“生物醫(yī)學(xué)新技術(shù)”范疇
《條例》共7章58條,旨在規(guī)范生物醫(yī)學(xué)新技術(shù)的臨床研究和臨床轉(zhuǎn)化應(yīng)用,促進(jìn)醫(yī)學(xué)科技創(chuàng)新,同時保障醫(yī)療質(zhì)量安全,維護(hù)人的尊嚴(yán)和健康。
《條例》第三條重點(diǎn)明確了“生物醫(yī)學(xué)新技術(shù)”的定義,是指“運(yùn)用生物學(xué)原理、作用于人體細(xì)胞或分子水平、我國境內(nèi)尚未臨床應(yīng)用的醫(yī)學(xué)專業(yè)手段或措施”。
具體而言,以下四類活動被《條例》界定為“生物醫(yī)學(xué)新技術(shù)臨床研究”:
-
直接對人體進(jìn)行操作;
-
對離體的細(xì)胞、組織、器官等進(jìn)行操作后,再植入或輸入人體;
-
對人的生殖細(xì)胞、合子、胚胎進(jìn)行操作后植入人體使其發(fā)育;
-
以及國家衛(wèi)生健康部門規(guī)定的其他方式。
過去,由于缺乏明確定義,一些技術(shù)形態(tài)難以歸類,監(jiān)管上出現(xiàn)模糊地帶。例如類器官等產(chǎn)品既不同于傳統(tǒng)藥品也非典型醫(yī)療器械,企業(yè)往往困惑于應(yīng)按照哪種路徑申報(bào)。《條例》通過定義賦予“生物醫(yī)學(xué)新技術(shù)”獨(dú)立法律地位,等于在藥品、器械之外開辟出第三類監(jiān)管軌道,與之各有側(cè)重又相互銜接。
03 擴(kuò)大臨床主體范圍:高校藥企可發(fā)起,三甲醫(yī)院實(shí)施
在舊有規(guī)制下,新技術(shù)臨床研究通常由醫(yī)療機(jī)構(gòu)主導(dǎo)進(jìn)行。例如2015年干細(xì)胞研究辦法就構(gòu)建了“以醫(yī)療機(jī)構(gòu)為責(zé)任主體”的管理模式。這樣的限定在一定程度上限制了企業(yè)和高校的直接參與。《條例》則明確拓寬了研究發(fā)起主體的范圍,為在我國境內(nèi)依法成立的法人單位。這意味著高校科研院所、生物醫(yī)藥企業(yè)等均被允許直接發(fā)起臨床研究項(xiàng)目,參與創(chuàng)新研發(fā)的早期探索。
與此同時,新規(guī)對臨床研究實(shí)施機(jī)構(gòu)設(shè)定了嚴(yán)格資質(zhì)要求:
-
機(jī)構(gòu)級別:須為三級甲等醫(yī)療機(jī)構(gòu);
-
倫理與學(xué)術(shù):擁有符合要求的臨床研究學(xué)術(shù)委員會和倫理委員會;
-
設(shè)施能力:具備與擬開展研究相適應(yīng)的資質(zhì)、場所設(shè)施、設(shè)備、專業(yè)人才和研究能力;
-
管理制度:建立保障研究質(zhì)量安全、符合倫理原則及受試者權(quán)益的管理制度;
-
經(jīng)費(fèi)保障:有穩(wěn)定充足的研究經(jīng)費(fèi)來源。
這種設(shè)計(jì)鼓勵了產(chǎn)學(xué)研醫(yī)協(xié)作:企業(yè)、高校的創(chuàng)新成果可以攜手醫(yī)院的臨床資源,加速推進(jìn)研究落地。而衛(wèi)生主管部門則通過備案和評估對全過程進(jìn)行指導(dǎo)監(jiān)管,形成各司其職又緊密協(xié)同的新格局。
此外,對企業(yè)而言,這提供了另一條商業(yè)化途徑——“院內(nèi)應(yīng)用”模式。企業(yè)可以專注于類器官等生物醫(yī)學(xué)新技術(shù)的研發(fā)和標(biāo)準(zhǔn)化,與醫(yī)院合作完成臨床研究;一旦衛(wèi)健委批準(zhǔn)應(yīng)用,企業(yè)可通過向醫(yī)院提供類器官培養(yǎng)技術(shù)服務(wù)、專用培養(yǎng)設(shè)備和試劑等方式,實(shí)現(xiàn)成果轉(zhuǎn)化和營利。這種模式下,企業(yè)不需要經(jīng)過藥監(jiān)部門繁瑣的上市審批,就能讓產(chǎn)品進(jìn)入臨床使用,大大縮短周期。當(dāng)然,如果類器官技術(shù)最終演變?yōu)闃?biāo)準(zhǔn)化產(chǎn)品(如通用型類器官移植物),企業(yè)仍可以選擇申報(bào)藥品/器械批件后更大規(guī)模推廣。無論哪種方式,《條例》的框架使得路線選擇一目了然,企業(yè)可根據(jù)自身技術(shù)特點(diǎn)和市場策略靈活規(guī)劃,而不再因政策模糊而舉棋不定。
04 明確審批方式,加速臨床轉(zhuǎn)化
備案+審批新模式
長期以來,新醫(yī)療技術(shù)從研究走向臨床常面臨審批周期長、流程不透明的問題。舊機(jī)制下,不僅需要多層行政審查,而且缺乏明確的辦結(jié)時限,影響了患者及時獲益。
本次《條例》針對此做出了重大優(yōu)化,建立了“備案+審批”相結(jié)合的高效監(jiān)管模式。在臨床研究階段,改為機(jī)構(gòu)自行學(xué)術(shù)和倫理審查通過后向國家平臺備案即可啟動研究,區(qū)別于此前2015年《干細(xì)胞臨床研究管理辦法(試行)》采用的事前行政審批,也為此前缺乏法定審批或備案渠道的大量不屬于干細(xì)胞、也不屬于藥品/器械的其它新技術(shù)——如某些細(xì)胞與基因治療(CGT)、類器官治療等——提供了合規(guī)路徑。在此基礎(chǔ)上,國家衛(wèi)健部門把準(zhǔn)入決策權(quán)下放給熟悉一線情況的機(jī)構(gòu),自己則重點(diǎn)加強(qiáng)過程中的監(jiān)督評估和風(fēng)險處置。這種備案制使得合規(guī)研究可以“先行先試”,大大提高了科研效率。
打通臨床研究到轉(zhuǎn)化應(yīng)用全鏈條
過往政策中,生物醫(yī)學(xué)新技術(shù)往往僅被允許開展臨床研究,但研究成果無法直接服務(wù)患者,臨床應(yīng)用路徑不清晰。例如干細(xì)胞研究試點(diǎn)曾明確規(guī)定,完成研究后不得直接用于臨床應(yīng)用,需轉(zhuǎn)入藥品注冊等途徑。《條例》針對這一痛點(diǎn)作出制度創(chuàng)新,不再局限于“臨床研究”階段,而是打通了從研究到應(yīng)用的完整轉(zhuǎn)化通道。
根據(jù)新規(guī),生物醫(yī)學(xué)新技術(shù)在臨床研究證明安全有效、符合倫理原則后,可直接向國家衛(wèi)生健康主管部門申請轉(zhuǎn)化為臨床應(yīng)用,經(jīng)審批即可用于患者。該流程類似于藥物的臨床試驗(yàn)申請(IND),但相對快速簡化,尤其適用于個性化、自體類器官療法等小批量創(chuàng)新。
臨床轉(zhuǎn)化雙軌并行
《條例》還開創(chuàng)了臨床轉(zhuǎn)化“雙軌制”:新技術(shù)成果既可以選擇傳統(tǒng)藥品器械注冊路徑,也可以依據(jù)本條例走技術(shù)轉(zhuǎn)化應(yīng)用路徑,兩條道路并行選擇。這一設(shè)計(jì)為細(xì)胞治療、基因療法等前沿產(chǎn)品提供了更靈活快速的落地渠道,真正實(shí)現(xiàn)科研創(chuàng)新與臨床應(yīng)用之間的無縫銜接。
舉例而言,類器官(organoids)是指利用成體干細(xì)胞或多能干細(xì)胞在體外三維培養(yǎng)形成的、具有一定結(jié)構(gòu)和功能的“微型器官”組織體。相較傳統(tǒng)二維細(xì)胞培養(yǎng),三維類器官更好地模擬了真實(shí)器官的功能與微觀結(jié)構(gòu),能夠長期穩(wěn)定傳代,是強(qiáng)大的疾病模型和新藥篩選工具。根據(jù)上述第二條定義,亦屬于本次《條例》列出的“生物醫(yī)學(xué)新技術(shù)臨床研究”范疇。
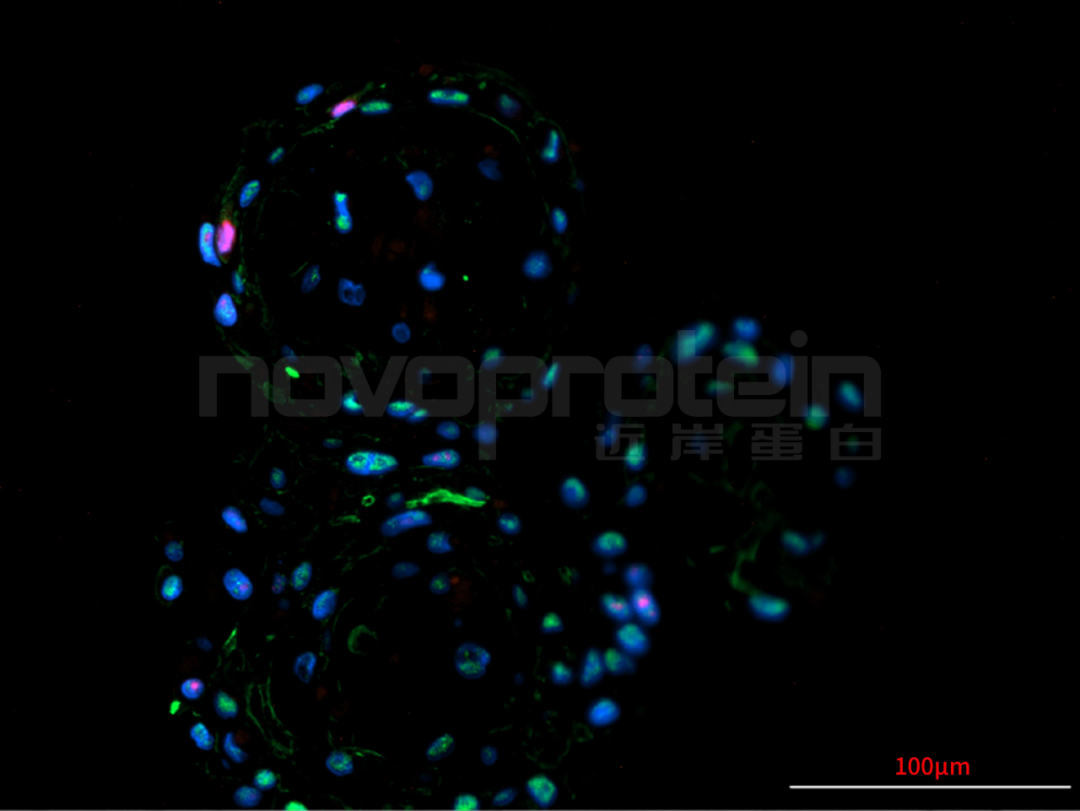
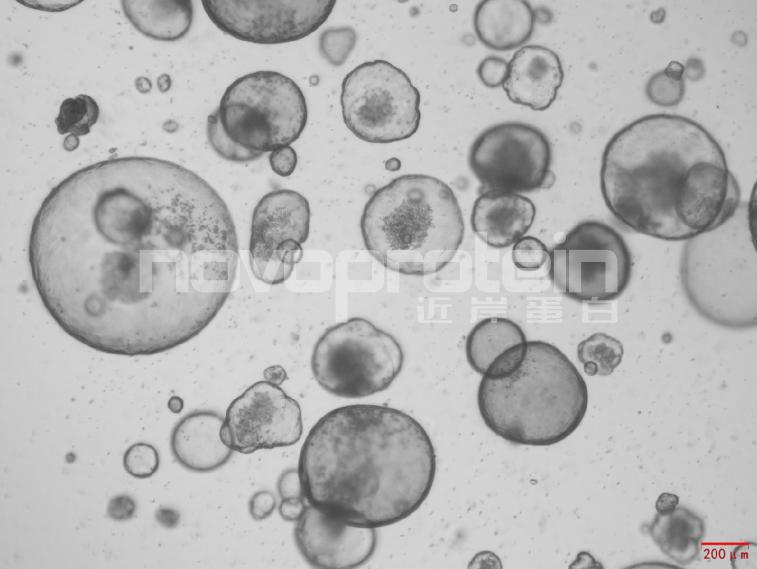
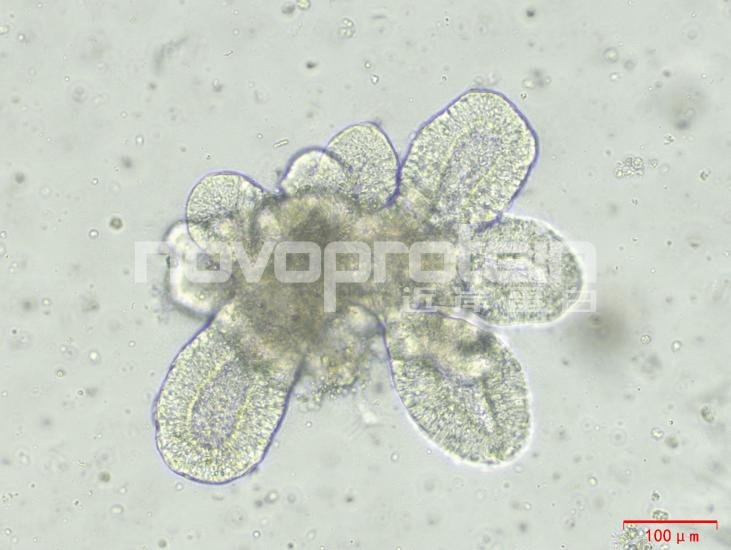
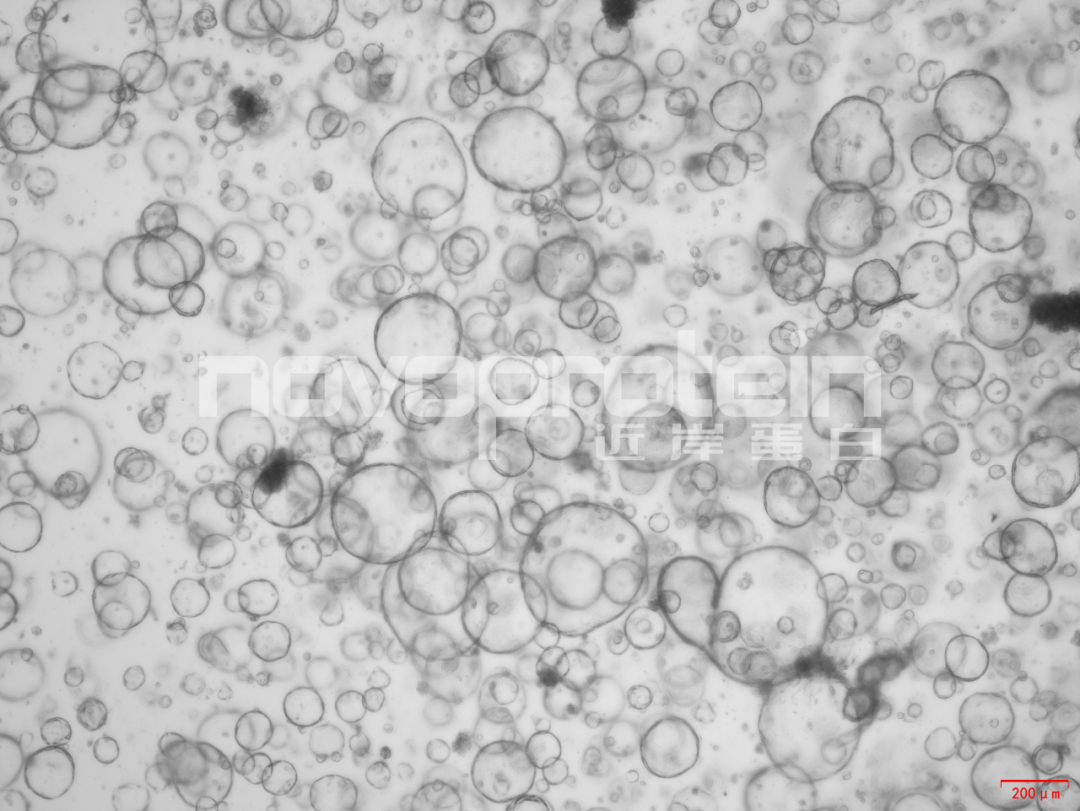
圖:經(jīng)近岸蛋白細(xì)胞因子驗(yàn)證培養(yǎng)的類器官(從左至右,從上至下:乳腺癌類器官、ipsc來源的人小腸類器官、小鼠胃類器官及小鼠膽管類器官)
自2013年被《Science》雜志評為年度十大科學(xué)突破以來,類器官技術(shù)發(fā)展突飛猛進(jìn),目前已廣泛應(yīng)用于癌癥、罕見病、神經(jīng)退行性疾病、感染等領(lǐng)域的研究。它不僅可用于模擬疾病機(jī)制、篩選藥物,還在個性化醫(yī)療方面展現(xiàn)出獨(dú)特價值——例如從癌癥患者腫瘤組織培養(yǎng)“腫瘤類器官”,在實(shí)驗(yàn)室測試多種藥物敏感性,從而為患者甄選出最有效的治療方案,被形象地稱為為患者“替身試藥”,有望大幅提升精準(zhǔn)治療效果。
盡管類器官在科研和藥物開發(fā)中大放異彩,其真正走向臨床應(yīng)用仍面臨諸多挑戰(zhàn)。在再生醫(yī)學(xué)領(lǐng)域,類器官有望用于替代或修復(fù)損傷組織(如培育血管類器官植入修復(fù)血管損傷),但此前缺少明確的臨床研究許可渠道。醫(yī)院和研究者若想嘗試將類器官用于患者(無論是直接移植,還是作為藥物療效預(yù)測的診斷技術(shù)),往往找不到適用的監(jiān)管規(guī)范,只能徘徊在研究階段。
現(xiàn)在,《條例》提供了清晰的路徑:將類器官相關(guān)創(chuàng)新納入“生物醫(yī)學(xué)新技術(shù)”范疇,只要不屬于法律明令禁止或重大倫理問題,就可以按照條例要求開展臨床研究和申請臨床轉(zhuǎn)化備案。類器官技術(shù)通常是在體外培養(yǎng)患者自體或同源細(xì)胞形成組織,再植回體內(nèi)或用于體外診療,從倫理和風(fēng)險角度看,相對于修改人類胚胎基因等高風(fēng)險技術(shù),要溫和、安全得多。
此前《條例》征求意見稿曾提出按風(fēng)險等級分級管理的思路:中低風(fēng)險的新技術(shù)臨床研究由省級衛(wèi)健部門審批,高風(fēng)險項(xiàng)目則報(bào)國家審批。雖然正式條文未明示分級,但可以預(yù)見在未來配套細(xì)則中會對技術(shù)風(fēng)險分層管理。類器官有望被歸入“中低風(fēng)險”序列,因其不涉及人類生殖系基因改造,倫理敏感性相對較低,安全性在動物試驗(yàn)中也更易驗(yàn)證。這意味著類器官相關(guān)臨床研究門檻相對較低,甚至可能由省級層面即可啟動,審批流程更為簡捷。如果研究結(jié)果良好,再報(bào)國家衛(wèi)健委審批進(jìn)入臨床應(yīng)用即可。這種模式無疑將加快類器官療法從實(shí)驗(yàn)室走向臨床的步伐。
05 監(jiān)管轉(zhuǎn)型提速,提升效能
監(jiān)管部門
具體而言,根據(jù)《條例》,生物醫(yī)學(xué)新技術(shù)臨床研究和轉(zhuǎn)化應(yīng)用由國家衛(wèi)健委主管,全國衛(wèi)生健康部門負(fù)責(zé)監(jiān)督管理。地方各級衛(wèi)生健康部門在屬地負(fù)責(zé)日常監(jiān)督執(zhí)法,其他相關(guān)部門在各自職責(zé)內(nèi)協(xié)同監(jiān)管。也就是說,此類新技術(shù)的準(zhǔn)入主要走“衛(wèi)生”系統(tǒng)而非傳統(tǒng)藥監(jiān)系統(tǒng),突出醫(yī)療專業(yè)監(jiān)管特點(diǎn)。
值得注意的是,中國生物技術(shù)發(fā)展中心副主任沈建忠在相關(guān)解讀中提到,條例授權(quán)國務(wù)院衛(wèi)生健康部門會同藥品監(jiān)管部門制定和調(diào)整生物醫(yī)學(xué)新技術(shù)與藥品、醫(yī)療器械的界定指導(dǎo)原則。這意味著未來將更清晰地區(qū)分哪些屬于醫(yī)療技術(shù)路徑、哪些屬于藥品或器械路徑,從而為新技術(shù)轉(zhuǎn)化路徑提供指引。
至于衛(wèi)健委與藥監(jiān)局如何劃分監(jiān)管邊界?目前尚待進(jìn)一步觀察。據(jù)財(cái)新網(wǎng)消息,一位已有產(chǎn)品上市的細(xì)胞治療公司創(chuàng)始人表示,《條例》所指的生物醫(yī)學(xué)新技術(shù),很可能是器官移植、骨髓移植等本就由衛(wèi)健委監(jiān)管的項(xiàng)目,“(關(guān)于具體項(xiàng)目),我相信兩個部門之間肯定會做一些界定”。
統(tǒng)一審批制度
同時,新規(guī)將原本存在的省級初審環(huán)節(jié)取消,避免了重復(fù)審核和地方標(biāo)準(zhǔn)不一的拖延,實(shí)現(xiàn)全國“一盤棋”的審批流程。這種提速增效的審批機(jī)制,體現(xiàn)出監(jiān)管部門“放管結(jié)合”的思路:在保障安全的前提下盡可能加快創(chuàng)新技術(shù)惠及患者的步伐。
加速審批速度
到了成果轉(zhuǎn)化審批環(huán)節(jié),更實(shí)行了嚴(yán)格的時限規(guī)定:國家衛(wèi)健委受理申請后,在5個工作日內(nèi)委托專業(yè)機(jī)構(gòu)完成技術(shù)和倫理評估,并在收到評估意見起15個工作日內(nèi)作出審批決定。法定流程不超過20個工作日,大幅縮短于舊規(guī)制下數(shù)月乃至數(shù)年的等待期。
特殊綠色通道
最后,《條例》還設(shè)置了特殊情形下的綠色通道和緊急應(yīng)用機(jī)制:對于治療危及生命且無有效手段的疾病或突發(fā)公共衛(wèi)生急需的新技術(shù),可“優(yōu)先審查審批”,在特別重大緊急情況下經(jīng)過嚴(yán)格論證可應(yīng)急使用尚在研究中的技術(shù)。這些舉措體現(xiàn)了監(jiān)管的靈活性與對重大臨床需求的積極響應(yīng)。
06 嚴(yán)守安全底線、保護(hù)患者權(quán)益
法律責(zé)任
在為創(chuàng)新“松綁”的同時,《條例》也通過完善退出和處罰機(jī)制為新技術(shù)應(yīng)用“加上安全帶”。
相比舊規(guī)制中法律責(zé)任模糊、震懾力不足的情況,新條例對違法違規(guī)行為設(shè)定了嚴(yán)格的法律責(zé)任條款。例如,對違規(guī)開展研究或應(yīng)用的機(jī)構(gòu)和責(zé)任人,監(jiān)管部門可處以高額罰款,情節(jié)嚴(yán)重者吊銷執(zhí)業(yè)資質(zhì),并可處以10年直至終身禁止從業(yè)的嚴(yán)厲處罰。如此大力度的懲戒在以往相關(guān)規(guī)定中尚屬首次。
同時,《條例》還建立了動態(tài)退出機(jī)制:即使某項(xiàng)新技術(shù)已獲準(zhǔn)臨床應(yīng)用,主管部門仍將根據(jù)證據(jù)對其進(jìn)行再評估;一旦發(fā)現(xiàn)安全性、有效性無法保證,可立即中止其臨床應(yīng)用。這一規(guī)定確保了“有進(jìn)有出”,對技術(shù)風(fēng)險始終保持警惕和控制。
受試者保護(hù)、倫理規(guī)范及收費(fèi)標(biāo)準(zhǔn)
新條例突出對參與臨床研究的受試者合法權(quán)益保護(hù),明確多項(xiàng)嚴(yán)格要求,強(qiáng)調(diào)全過程的風(fēng)險管理和受試者保護(hù):要求臨床研究機(jī)構(gòu)嚴(yán)格按備案方案執(zhí)行研究,落實(shí)風(fēng)險預(yù)防和處置措施,研究結(jié)束后對受試者進(jìn)行長期隨訪,評估技術(shù)的長期安全性和有效性;研究記錄和原始數(shù)據(jù)需至少保存30年,涉及人類后代影響的需保存。
這些舉措在舊制度中未有明確要求,如今作為硬性規(guī)定寫入《條例》,為受試者提供了貫穿研究全程的保障,大大提高了監(jiān)管的剛性和力度。可以說,新規(guī)在鼓勵大膽創(chuàng)新的同時,也筑牢了安全和倫理的紅線,確保行業(yè)發(fā)展不偏離保障人民健康權(quán)益的初心。
在此基礎(chǔ)上,過去委員會擔(dān)心類器官、CGT等生物醫(yī)學(xué)新技術(shù)項(xiàng)目缺乏政策支持而不敢批準(zhǔn)的情況將大為改善——只要符合條件、資料齊備,倫理審查和備案變得有章可循、透明高效。這實(shí)際上降低了合規(guī)成本和不確定性。同時,知情同意流程的規(guī)范確保受試患者充分了解參與例如“類器官移植試驗(yàn)”可能的風(fēng)險和收益,這既保護(hù)患者權(quán)利,也減少了項(xiàng)目后顧之憂。因此,類器官團(tuán)隊(duì)可以更放心地開展創(chuàng)新性臨床探索,在遵守法規(guī)前提下勇于嘗試。
同樣值得關(guān)注的是,《意見稿》明確就醫(yī)療機(jī)構(gòu)不得以任何形式向受試者收取與研究內(nèi)容相關(guān)的任何費(fèi)用做出了規(guī)定。在其批準(zhǔn)進(jìn)入臨床應(yīng)用后,經(jīng)省級人民政府醫(yī)療價格主管部門會同衛(wèi)生主管部門納入醫(yī)療服務(wù)價格項(xiàng)目并確定收費(fèi)標(biāo)準(zhǔn)。而《條例》規(guī)定,生物醫(yī)學(xué)新技術(shù)臨床研究結(jié)束后可以向國務(wù)院衛(wèi)生健康部門申請轉(zhuǎn)化應(yīng)用,審批通過的醫(yī)療機(jī)構(gòu)開展臨床應(yīng)用可以按照規(guī)定收取費(fèi)用。
07 一錘定音,全面開花 有法可依,加速前進(jìn)
從地方試點(diǎn)到全國“基本法”
近年海南博鰲樂城國際醫(yī)療旅游先行區(qū)在細(xì)胞治療、基因療法等新技術(shù)臨床應(yīng)用方面進(jìn)行了大膽探索,積累了寶貴經(jīng)驗(yàn)。例如樂城先行區(qū)通過特許準(zhǔn)入和真實(shí)世界研究,率先引進(jìn)應(yīng)用了一批境內(nèi)外前沿藥械和生物技術(shù),為監(jiān)管創(chuàng)新提供了試驗(yàn)田和樣板。
然而此前這些創(chuàng)新舉措主要局限于樂城園區(qū)內(nèi),未能在更大范圍復(fù)制推廣。《條例》的出臺意味著將樂城等先行先試地區(qū)的經(jīng)驗(yàn)做法上升為國家層面的制度設(shè)計(jì),在全國范圍內(nèi)建立起生物醫(yī)學(xué)新技術(shù)創(chuàng)新應(yīng)用的推廣機(jī)制。
可以預(yù)見,原本只有在樂城先行區(qū)才能接受的尖端療法,未來有望在北京、上海等地的醫(yī)院同步應(yīng)用于患者,實(shí)現(xiàn)政策紅利惠及全國。對于生物醫(yī)藥產(chǎn)業(yè)而言,這無疑釋放出積極信號:只要遵循規(guī)范、確保安全,新技術(shù)創(chuàng)新不再受制于狹小的試驗(yàn)范圍,而是擁有廣闊的市場應(yīng)用前景。這將激勵更多企業(yè)和研究機(jī)構(gòu)投入前沿技術(shù)研發(fā),推動我國生物醫(yī)學(xué)創(chuàng)新成果加速轉(zhuǎn)化為臨床生產(chǎn)力。
法規(guī)層級躍升:從部門規(guī)章到國務(wù)院條例
《條例》的頒布標(biāo)志著我國對生物醫(yī)學(xué)新技術(shù)監(jiān)管從部門規(guī)章上升為國務(wù)院行政法規(guī),制度權(quán)威性顯著提升。
回顧以往,我國曾通過部門規(guī)章探索新技術(shù)監(jiān)管路徑:例如2015年原國家衛(wèi)計(jì)委和原食藥監(jiān)總局聯(lián)合發(fā)布的《干細(xì)胞臨床研究管理辦法(試行)》,以醫(yī)療機(jī)構(gòu)為責(zé)任主體開展干細(xì)胞臨床研究,開啟了干細(xì)胞研究規(guī)范管理的先河。此后2018年國家衛(wèi)健委發(fā)布了《醫(yī)療技術(shù)臨床應(yīng)用管理辦法》(委令第1號),通過負(fù)面清單方式對醫(yī)療新技術(shù)臨床應(yīng)用實(shí)行分類管理,對高風(fēng)險技術(shù)嚴(yán)格管控。然而,這些措施屬于部門規(guī)章或規(guī)范性文件,法律效力和覆蓋范圍有限。
與此同時,近年來,從美國FDA到中國NMPA,監(jiān)管機(jī)構(gòu)相繼調(diào)整政策,逐步接納類器官等生物醫(yī)學(xué)新技術(shù)進(jìn)入主流監(jiān)管體系:美國出臺《FDA現(xiàn)代化法案2.0》允許以類器官等替代動物實(shí)驗(yàn),中國NMPA在2025年發(fā)布的罕見病藥物試驗(yàn)指導(dǎo)原則中首次將類器官數(shù)據(jù)納入藥物研發(fā)證據(jù)鏈。這些舉措表明,以類器官為代表的生物醫(yī)學(xué)新技術(shù)正日益得到官方認(rèn)可,被視為未來科技創(chuàng)新的重要支撐。
如今《條例》以行政法規(guī)形式由國務(wù)院統(tǒng)一出臺,意味著對生物醫(yī)學(xué)新技術(shù)的管理有了更高層級的法律保障和全國統(tǒng)一的規(guī)范,在制度權(quán)威性和執(zhí)行力度上都大大增強(qiáng)。
08 總結(jié)
綜上,新條例的核心亮點(diǎn)可以歸納為五個方面。
首先是明確定義了“生物醫(yī)學(xué)新技術(shù)”的法律邊界,首次將作用于人體細(xì)胞、分子水平、尚未臨床應(yīng)用的創(chuàng)新手段納入獨(dú)立監(jiān)管體系,區(qū)別于傳統(tǒng)藥品和醫(yī)療器械管理路徑,為干細(xì)胞、基因編輯、類器官等前沿方向確立了合法身份。
其次是擴(kuò)大了創(chuàng)新主體的范圍。過去臨床研究多由醫(yī)院單獨(dú)承擔(dān),如今條例允許高校、科研院所、企業(yè)等法人單位發(fā)起項(xiàng)目,三甲醫(yī)院作為實(shí)施機(jī)構(gòu),共同構(gòu)建產(chǎn)學(xué)研醫(yī)協(xié)同創(chuàng)新格局。
第三是加速了臨床轉(zhuǎn)化流程。新規(guī)實(shí)現(xiàn)備案與審批并行,形成“研究—評估—應(yīng)用”閉環(huán);審批時限縮短至20個工作日,較舊規(guī)制下需數(shù)月報(bào)批的周期顯著提速,使科研成果更快惠及患者。
第四是打通了審批通道與轉(zhuǎn)化路徑。新條例構(gòu)建了與藥品、器械并行的“第三條合規(guī)通路”,研究成果可在完成安全驗(yàn)證后直接申請臨床應(yīng)用,不再陷入以往“研究能做、臨床不能用”的尷尬狀態(tài)。
第五是監(jiān)管體系更加完備。法規(guī)從備案、倫理、評估到退出、處罰形成全鏈條機(jī)制,對違規(guī)行為設(shè)立高額罰款與從業(yè)禁令,既保障了受試者安全,也樹立了行業(yè)底線。
總的來看,這部以國務(wù)院行政法規(guī)形式頒布的《生物醫(yī)學(xué)新技術(shù)條例》,不僅在制度層級上實(shí)現(xiàn)了從“部委規(guī)章”到“國家法規(guī)”的跨越,也意味著監(jiān)管體系從局部試點(diǎn)邁向全國統(tǒng)一執(zhí)行。它所釋放的信號十分明確——生物醫(yī)學(xué)創(chuàng)新正在進(jìn)入法治化、體系化、加速化的新階段,一個更清晰、更開放、更具確定性的創(chuàng)新賽道,正全面展開。
近岸蛋白相關(guān)產(chǎn)品
近岸蛋白依靠專業(yè)的研發(fā)團(tuán)隊(duì),精心打造類器官驗(yàn)證平臺,可提供一系列經(jīng)驗(yàn)證、高活性、高穩(wěn)定性的類器官培養(yǎng)產(chǎn)品,助您高效快速開展類器官實(shí)驗(yàn)!
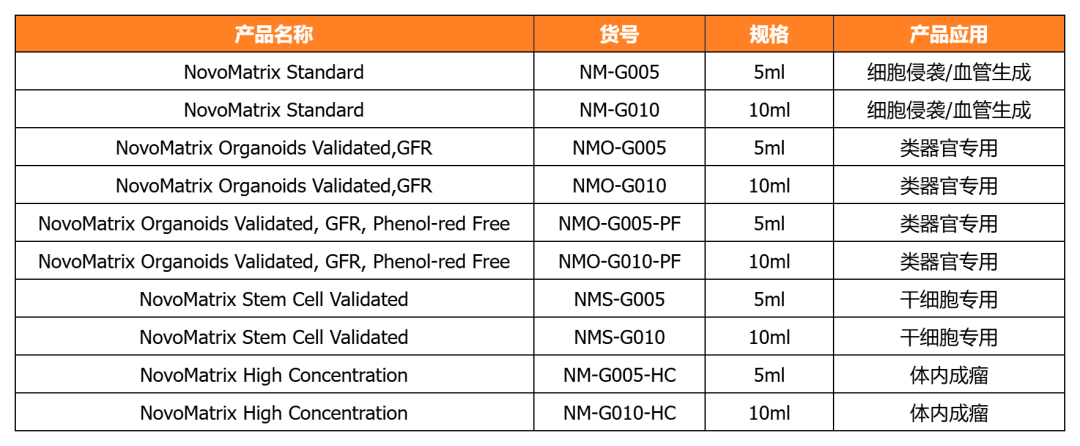
NovoMatrix-經(jīng)類器官培養(yǎng)驗(yàn)證的基質(zhì)膠
近岸蛋白提供經(jīng)類器官/干細(xì)胞培養(yǎng)驗(yàn)證的系列NovoMatrix基質(zhì)膠,產(chǎn)品批次穩(wěn)定可控,讓您的類器官/干細(xì)胞培養(yǎng)更可控!
NovoMatrix支持多種類器官培養(yǎng)
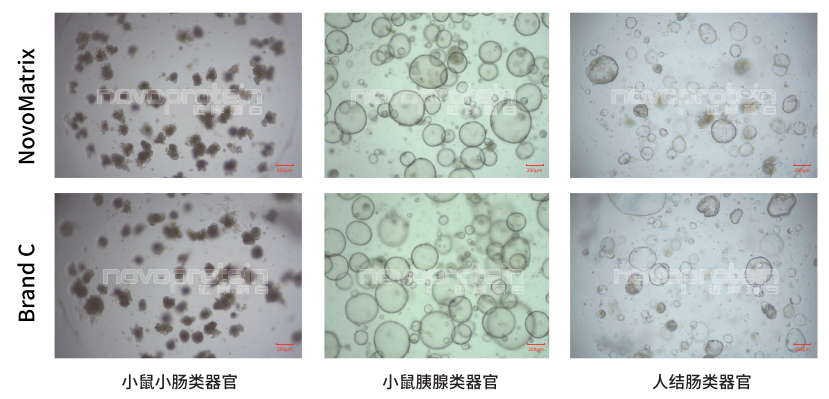
NovoMatrix Organoid Validated,GFR,Phenol Red-Free (Cat.No.:NMO-G005-PF) and a similar product of Brand C were used for culture of different organoid models
NovoMatrix支持類器官穩(wěn)定傳代

NovoMatrix Organoid Validated,GFR,Phenol Red-Free (Cat.No.:NMO-G005-PF) maintain stable passage of organoids comparable to a similar product of Brand C
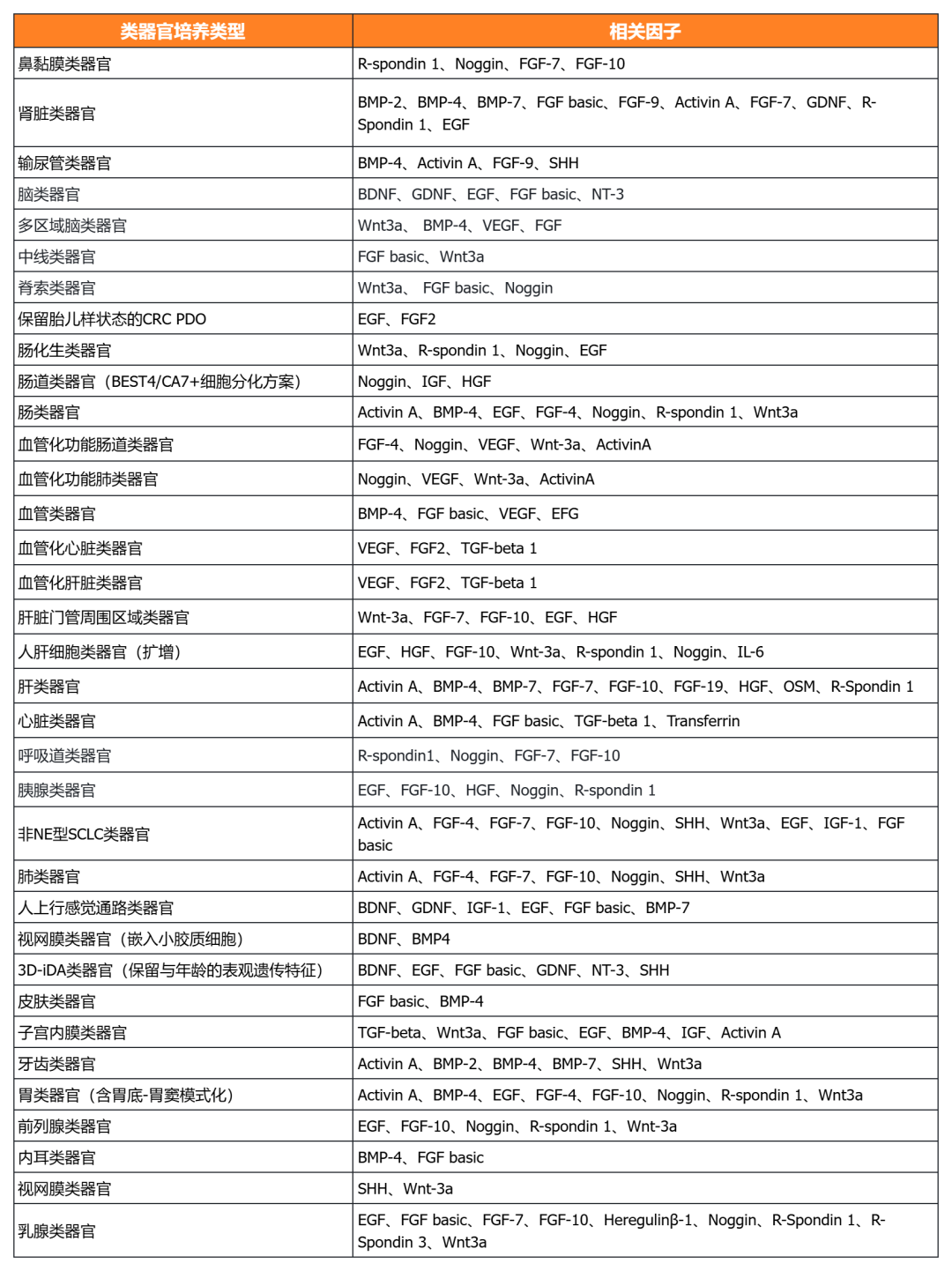
經(jīng)類器官培養(yǎng)驗(yàn)證的細(xì)胞因子
近岸蛋白自主研發(fā)生產(chǎn)并經(jīng)過類器官培養(yǎng)驗(yàn)證的低內(nèi)毒素Activin A、BMP-4、EGF、FGF-7、FGF-10、FGFb、FGF8b、HGF、IGF1、Noggin、NRG1、R-Spondin 1/3和Wnt3a等細(xì)胞因子,已經(jīng)在人腫瘤類器官如垂體瘤類器官、腦膜瘤類器官、乳腺癌類器官,人正常類器官如ipsc來源的人小腸類器官,以及小鼠正常類器官如小腸類器官、結(jié)腸類器官、胃類器官以及膽管類器官等體外模型上進(jìn)行了驗(yàn)證。
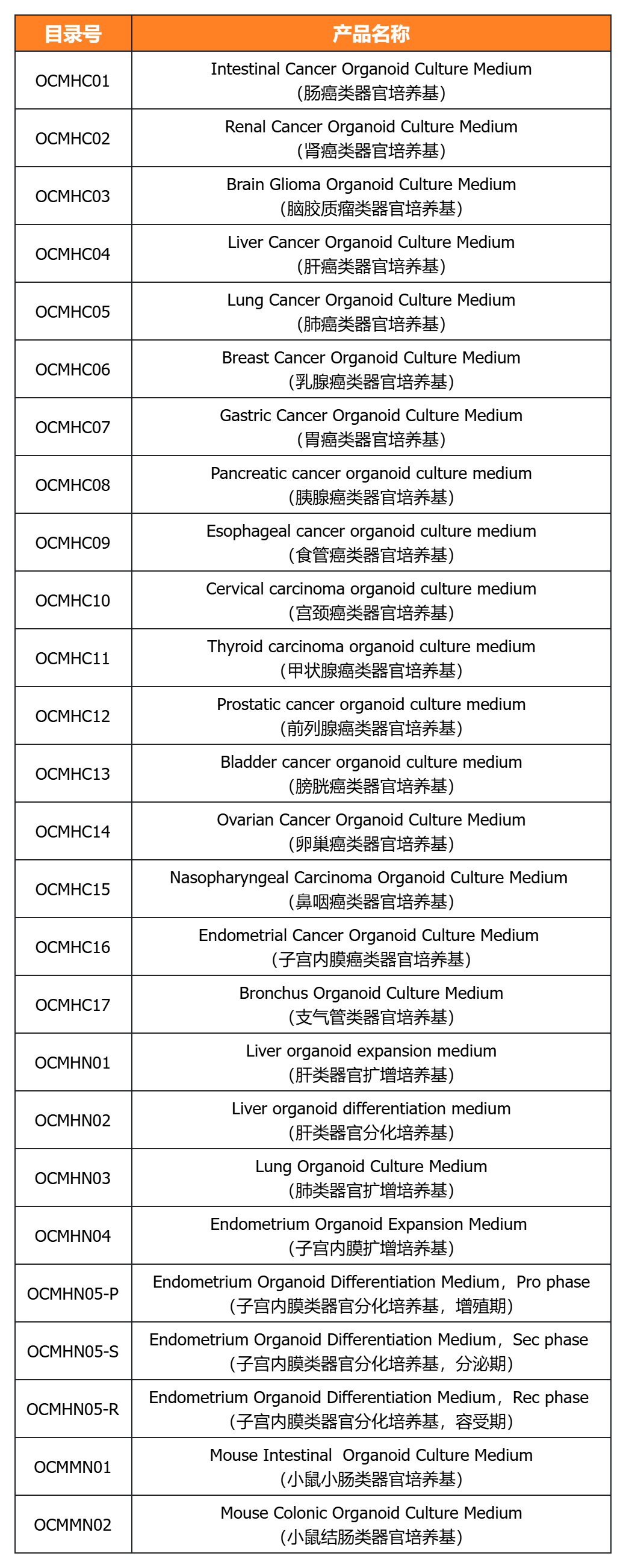
經(jīng)類器官培養(yǎng)驗(yàn)證的完全培養(yǎng)基