【導讀聚焦】
代謝重編程是生命活動與疾病發生的核心機制,腸道菌群代謝產物、膳食營養成分及神經-器官信號通路的交互作用,正逐步揭示疾病防治的全新靶點。近期發表于Nature, Nature metabolism, Advanced Science等頂刊的研究,從微生態、膳食代謝、神經調控等多個維度,解析了代謝分子在腫瘤、代謝性疾病、炎癥等領域的關鍵作用,本期納入了三篇果糖研究。
腸道菌群和代謝物展現出顯著的促腫瘤/抗腫瘤干預潛力。卷曲乳酸桿菌分泌的芥酸可通過PPAR-δ通路誘導宮頸癌鐵死亡,約氏乳桿菌則能合成鵝去氧膽酸降低高脂飲食誘導的結直腸癌風險;含糖飲料中的果糖與葡萄糖經SORD酶增強結直腸癌轉移,而果糖還可通過調控癌癥相關成纖維細胞與腫瘤細胞的交互驅動結直腸癌進展。
膳食成分的代謝機制解析為飲食-代謝性疾病的干預提供科學依據。適應膳食纖維的腸道菌群可清除膳食果糖、逆轉肝脂肪變性;N-乳酰苯丙氨酸通過抑制AgRP神經元減少小鼠攝食;7-酮基脫氧膽酸作為天然TGR5激動劑,能通過內質網鈣釋放促進結腸黏膜愈合,為潰瘍性結腸炎治療提供新靶點;杏仁核-肝臟信號通路可獨立于經典激素軸,協調應激狀態下的血糖反應,為慢性應激相關代謝異常提供了全新的干預方向。
【導讀目錄】
-
Advanced Science | 卷曲乳酸桿菌來源的芥酸介導PPAR-??通路誘導宮頸癌類器官鐵死亡
-
Advanced Science | 7-酮基脫氧膽酸通過TGR5-IP3R通路誘導內質網鈣釋放促進結腸黏膜愈合
-
Nature Metabolism | N-乳酰苯丙氨酸(Lac-Phe)抑制AgRP神經元誘導小鼠攝食減少
-
Nature Metabolism | 含糖飲料中的果糖和葡萄糖通過山梨糖醇脫氫酶(SORD)增強結直腸癌轉移
-
Cancer Research | 約氏乳桿菌合成鵝去氧膽酸以降低高脂飲食誘導結直腸癌的易感性
-
Nature Metabolism | 適應膳食纖維的腸道菌群能夠清除膳食中的果糖并逆轉肝脂肪變性
-
Gut | 果糖通過調控癌癥相關成纖維細胞與腫瘤細胞間的交互作用驅動結直腸癌進展
-
Nature | 杏仁核-肝臟信號通路協調應激下的血糖反應
【資源領取】
本期導讀文獻原文,請在公眾號后臺回復“2025年10月繪譜導讀”,即可獲取資源鏈接。
【壹】
Advanced Science | 卷曲乳酸桿菌來源的芥酸介導PPAR-??通路誘導宮頸癌類器官鐵死亡

陰道微生物群在女性生殖健康中扮演重要角色,但其在宮頸癌發生發展中的具體作用機制尚不明確。本研究通過16S rRNA測序發現,宮頸癌患者陰道菌群中乳酸桿菌豐度顯著降低,而卷曲乳酸桿菌(Lactobacillus crispatus)作為健康陰道狀態的代表菌種,其代謝產物芥酸(Erucic Acid, EA)能夠通過激活PPAR-δ信號通路,增強脂肪酸氧化(FAO),誘導大量活性氧(ROS)產生,最終引發宮頸癌類器官和細胞發生鐵死亡。該研究首次揭示了卷曲乳酸桿菌來源的芥酸在宮頸癌治療中的潛在作用,為宮頸癌的微生態治療提供了新思路。
1.首先,為探究宮頸癌患者與健康女性陰道菌群的差異,研究采用16S rRNA測序分析了23例宮頸癌患者和89例健康女性的陰道分泌物。結果顯示,宮頸癌患者陰道菌群中乳酸桿菌豐度顯著降低,而普雷沃菌、消化鏈球菌等機會致病菌增多。進一步從健康女性中分離出卷曲乳酸桿菌(LC 001),并通過熒光染色確認其良好活性。作為驗證,研究構建了來源于患者的宮頸癌類器官(PDO),并將其與無細胞上清液(CFS)共培養,發現CFS能顯著抑制類器官活力和生長,而菌體本身無此效應。在宮頸癌細胞系(Ca Ski、C-33A)和細胞源性異種移植(CDX)模型中,CFS同樣抑制了腫瘤增殖、遷移和侵襲,且該效應與pH值無關。
2.為解析CFS抑制腫瘤的機制,研究對CFS處理的類器官進行單細胞RNA測序。結果顯示,CFS處理組中與細胞死亡抵抗相關的細胞比例下降,而鐵死亡相關通路顯著富集。進一步實驗證實,CFS可誘導ROS積累和脂質過氧化,且該效應可被鐵死亡抑制劑Ferrostatin-1逆轉。隨后通過非靶向代謝組學分析發現,芥酸在CFS中含量顯著上升。體外實驗表明,芥酸可抑制宮頸癌細胞增殖、遷移和侵襲,并誘導GSH耗竭、MDA和4-HNE積累等鐵死亡特征。在CDX模型中,芥酸同樣表現出顯著的抑瘤效果。
3.最后,為闡明芥酸誘導鐵死亡的信號通路,機制研究發現芥酸作為PPAR-δ的配體,激活下游脂肪酸氧化關鍵酶(如ACOX1、CPT2),促進NADH和乙酰輔酶A生成,增強ROS產生。使用PPAR-δ拮抗劑GSK3787可逆轉芥酸誘導的ROS升高、鐵死亡及腫瘤抑制效應,證實PPAR-δ-FAO-ROS通路在其中的核心作用。
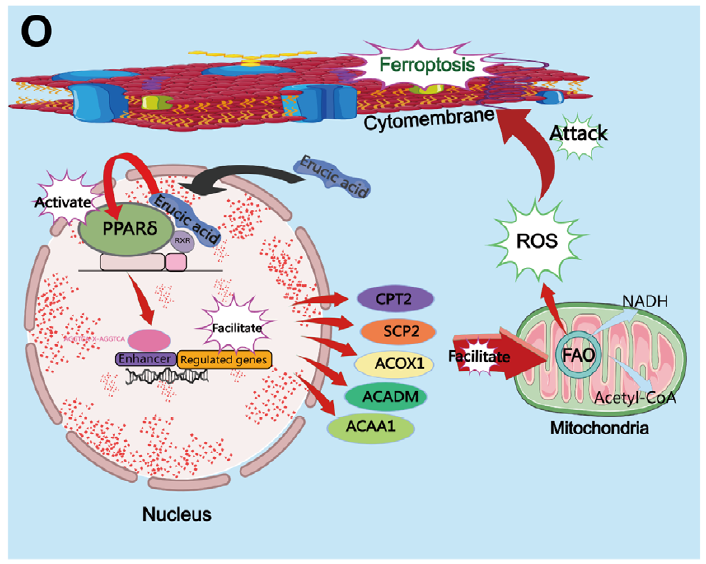
參考文獻:Zhen Q, Xu Y, Xu Y, et al. Erucic Acid, Derived by Lactobacillus Crispatus, Induces Ferroptosis in Cervical Cancer Organoids Through the PPAR-δ Signaling Pathway. Adv Sci. 2025
【貳】
Advanced Science | 7-酮基脫氧膽酸通過TGR5-IP3R通路誘導內質網鈣釋放促進結腸黏膜愈合

潰瘍性結腸炎(UC)的特征之一是結腸黏膜損傷后愈合障礙,而能夠促進黏膜修復的內源性代謝物尚不明確。本研究發現,UC患者和結腸炎小鼠血清及結腸組織中7-酮基脫氧膽酸(7-KDCA)水平顯著降低,且與疾病嚴重程度呈負相關,機制研究表明,7-KDCA通過激活TGR5受體,誘導內質網鈣離子釋放,促進腸上皮細胞遷移,從而加速黏膜愈合。該研究首次揭示了7-KDCA作為一種天然TGR5激動劑在結腸黏膜修復中的關鍵作用,為UC治療提供了新靶點。
1.首先,研究采用LC-MS靶向代謝組學技術分析了UC患者和健康對照者的血清樣本。結果顯示,7-KDCA、脫氧膽酸(DCA)和石膽酸(LCA)水平在UC患者中顯著降低,其中7-KDCA的降低與炎癥指標(如CRP、ProCT)負相關,與白蛋白正相關,提示其與疾病嚴重程度密切相關。在DSS誘導的結腸炎小鼠模型中,這三種膽汁酸在急性期顯著下降,恢復期回升,進一步驗證了其與結腸病理變化的相關性。
2.為評估7-KDCA對黏膜修復的促進作用,研究在DSS誘導和活檢損傷兩種小鼠模型中口服補充7-KDCA。結果顯示,7-KDCA能顯著提高小鼠存活率、減輕體重下降、降低疾病活動指數、改善結腸長度和通透性,并上調緊密連接蛋白(如ZO-1、claudin-1)的表達。內鏡和組織學分析進一步證實,7-KDCA治療組黏膜潰瘍和水腫明顯減輕,愈合效果優于DCA和LCA。
3.體外劃痕實驗和Transwell遷移實驗結果顯示,7-KDCA能顯著增強人結腸上皮細胞(NCM-460和HT-29)的遷移能力,而對細胞增殖影響不顯著。通過siRNA敲低和藥理學抑制實驗發現,7-KDCA的促遷移和愈合作用依賴于TGR5受體,而非FXR受體。進一步機制研究表明,7-KDCA激活TGR5后,通過cAMP-PKA信號通路上調IP3水平,激活IP3受體,誘導內質網鈣離子釋放,從而驅動細胞遷移。
4.最后,研究通過AAV-shRNA在體敲低TGR5或IP3R,驗證了7-KDCA的修復作用在TGR5或IP3R缺失小鼠中被完全阻斷,而在FXR敲低小鼠中仍有效。此外,藥代動力學分析顯示,口服7-KDCA后其在腸道中濃度高而血中濃度極低,未引起全身性副作用(如膽囊充盈),表明其具有良好的腸道限制性和安全性。

參考文獻:Zhang J, Jiang F, Xia W, et al. 7-Ketodeoxycholic Acid Promotes Colonic Mucosal Healing by Inducing Calcium Release from Endoplasmic Reticulum via the TGR5-IP3R Pathway. Adv Sci. 2025
【叁】
Nature Metabolism | N-乳酰苯丙氨酸(Lac-Phe)抑制AgRP神經元誘導小鼠攝食減少

N-乳酰苯丙氨酸(Lac-Phe)是一種乳酸鹽衍生的循環代謝產物,是劇烈運動后血漿中最顯著升高的代謝物,具有顯著的抗肥胖作用,但Lac-Phe引起的攝食減少的機制仍不清楚。本文揭示了Lac-Phe介導代謝改善的分子和神經生物學機制,并指出這種運動誘導的代謝物可能對多種人類疾病中具有潛在治療價值。
1.行為學與中樞作用驗證:給小鼠腹腔或腦室內注射Lac-Phe,發現其能顯著減少正常飲食和高脂飲食小鼠攝食行為。通過c-Fos 染色(神經元活動標志物)發現, Lac-Phe會激活小鼠腦干兩個與食欲調控密切相關的神經元-孤束核(NTS)和下丘腦室旁核(PVH)。進一步用TRAP技術抑制激活的NTS神經元,發現抑制NTS神經元不影響Lac-Phe的厭食效應,而抑制激活的PVH神經元則厭食效應減弱。
2.神經環路與神經元作用研究:腦片電生理實驗表明Lac-Phe不直接激活PVH神經元。通過狂犬病毒示蹤,發現PVH神經元接受來自下丘腦弓狀核(ARH)AgRP神經元的抑制性輸入。利用電生理、鈣成像、c-Fos檢測等多種技術,證實Lac-Phe可直接抑制AgRP神經元活性。且該抑制需AgRP神經元參與,激活AgRP神經元會阻斷Lac-Phe的攝食抑制作用。
3.分子機制探究:電生理實驗顯示Lac-Phe通過激活AgRP神經元上的ATP敏感性鉀(KATP)通道抑制其活動,藥理學阻斷或利用CRISPR-Cas9敲除KATP通道均顯著減弱Lac-Phe的攝食抑制作用,證實KATP通道是介導其神經抑制與代謝效應的關鍵分子。
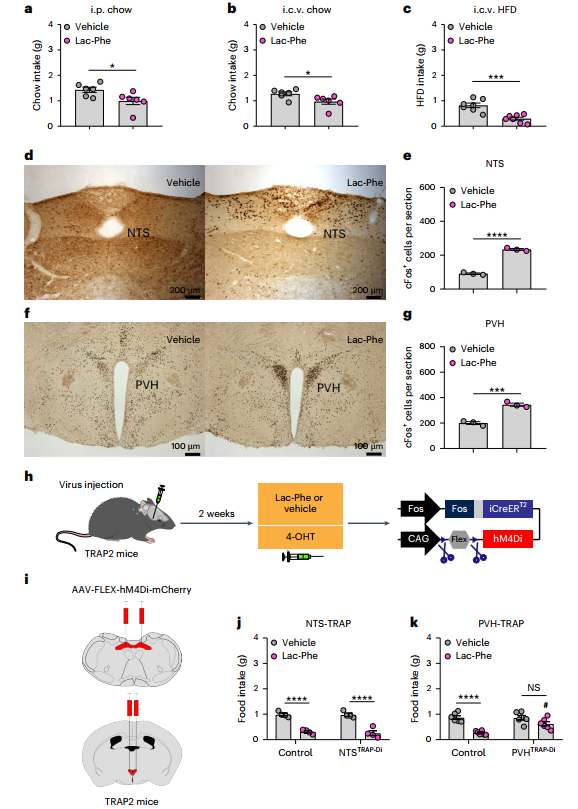
參考文獻:Liu H, et al. Lac-Phe induces hypophagia by inhibiting AgRP neurons in mice. Nature Metabolism. 2025
【肆】
Nature Metabolism | 含糖飲料中的果糖和葡萄糖通過山梨糖醇脫氫酶(SORD)增強結直腸癌轉移

含糖飲料(SSBs)含大量果糖與葡萄糖,已證實與結直腸癌(CRC)發病風險升高相關,但對CRC進展后期(如轉移)的影響及機制尚不明確。全球SSB消費量持續上升,且年輕人群CRC發病率與死亡率上升,部分CRC患者確診后仍飲用SSB。本研究旨在探究SSB中的果糖與葡萄糖是否促進晚期CRC轉移,并揭示其分子機制,為CRC患者的飲食建議與治療提供依據。
1.設置葡萄糖組、果糖組以及葡萄糖+果糖組(模擬SSB糖組成),評估其對CRC細胞生長、遷移和侵襲的影響。結果顯示,葡萄糖+果糖組顯著增強有遷移能力細胞系的遷移和侵襲。在CRC小鼠模型中,葡萄糖+果糖喂養組的肝轉移灶數量顯著增加。
2.代謝組學分析表明,葡萄糖+果糖組唯''一顯著升高的代謝物為山梨糖醇。同位素標記實驗證實,果糖可經SORD酶的逆向反應生成山梨糖醇,該過程消耗NADH并生成NAD+,葡萄糖則通過糖酵解提供NADH輔助該反應。SORD介導的逆向反應會導致細胞內的NAD+/NADH比值升高,從而會顯著增強糖酵解和三羧酸循環的活性,進而激活甲羥戊酸通路,使用他汀類藥物抑制甲羥戊酸通路可抑制癌細胞的遷移能力。
3.臨床CRC組織樣本中SORD表達高于正常組織,并且SORD的高表達主要集中在腫瘤上皮細胞,尤其是在那些具有干細胞特性的腫瘤細胞亞群中。CRISPR-Cas9敲除SORD顯著降低CRC細胞的遷移與侵襲能力,而調控NAD+/NADH比值升高可恢復SORD敲除細胞的遷移能力。
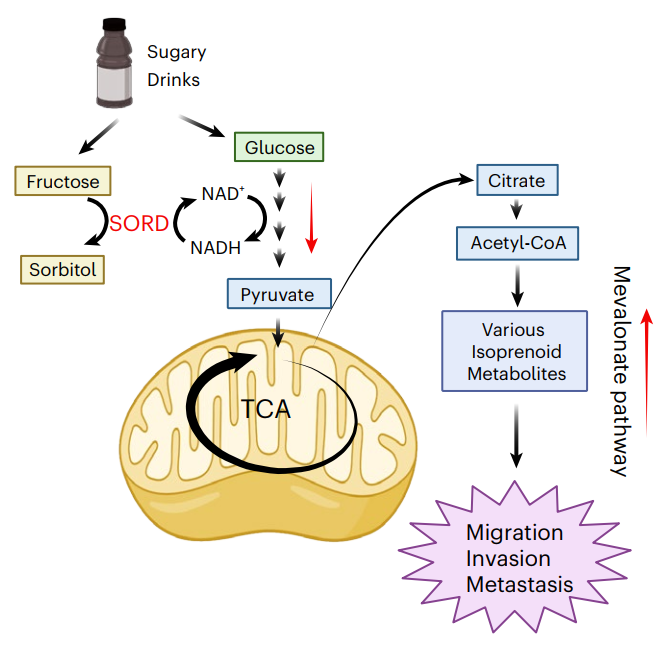
參考文獻:Feng T, et al. Fructose and glucose from sugary drinks enhance colorectal cancer metastasis via SORD. Nature Metabolism. 2025
【伍】
Cancer Research | 約氏乳桿菌合成鵝去氧膽酸以降低高脂飲食誘導結直腸癌的易感性

高脂肪飲食(HFD)與結直腸癌(CRC)呈正相關,但不同個體對HFD引發的促腫瘤作用的敏感性存在顯著差異。更深入理解調控高脂飲食效應的機制,有助于為結直腸癌的精準預防策略提供依據。本研究發現,L. johnsonii(約氏乳桿菌)的定植能夠增加HFD暴露小鼠腸道中的熊去氧膽酸(CDCA)水平,揭示了“約氏乳桿菌-鵝去氧膽酸”這一益生菌干預通路,為減少高脂飲食誘導的結直腸癌進展提供了潛在途徑。
1.對HIC-R(對結直腸癌具有抵抗力)和HIC-S(結直腸癌易感)小鼠的糞便樣本進行16S rRNA測序,結果發現,兩組小鼠的腸道菌群存在顯著差異。HIC-R小鼠腸道中乳桿菌屬(Lactobacillus)和雙歧桿菌屬(Bifidobacterium)等潛在益生菌豐度顯著升高,特別是約氏乳桿菌(Lactobacillus johnsonii)的富集尤為突出。
2.通過非靶向代謝組學分析初步發現,膽汁酸分泌和生物合成通路顯著富集。進一步通過靶向代謝組學發現,膽汁酸譜發生了顯著變化,腸道中CDCA的含量顯著增加。進一步給予L. johnsonii灌胃處理,顯著提高了模型小鼠糞便CDCA水平。約氏乳桿菌通過小鼠腸道中CDCA水平,進一步減少腫瘤數量和體積、抑制腫瘤生長和降低其異常增生。
3.對CDCA刺激后的腫瘤細胞團進行了RNA-Seq分析,結果發現,與線粒體翻譯和呼吸電子傳遞鏈徑相關的基因出現了顯著下調。
4.L. johnsonii通過膽鹽水解酶(BSH)將結合膽酸轉化為CDCA,并且CDCA誘導線粒體功能障礙和氧化應激,從而促進細胞凋亡,有效地抑制腫瘤的發展。
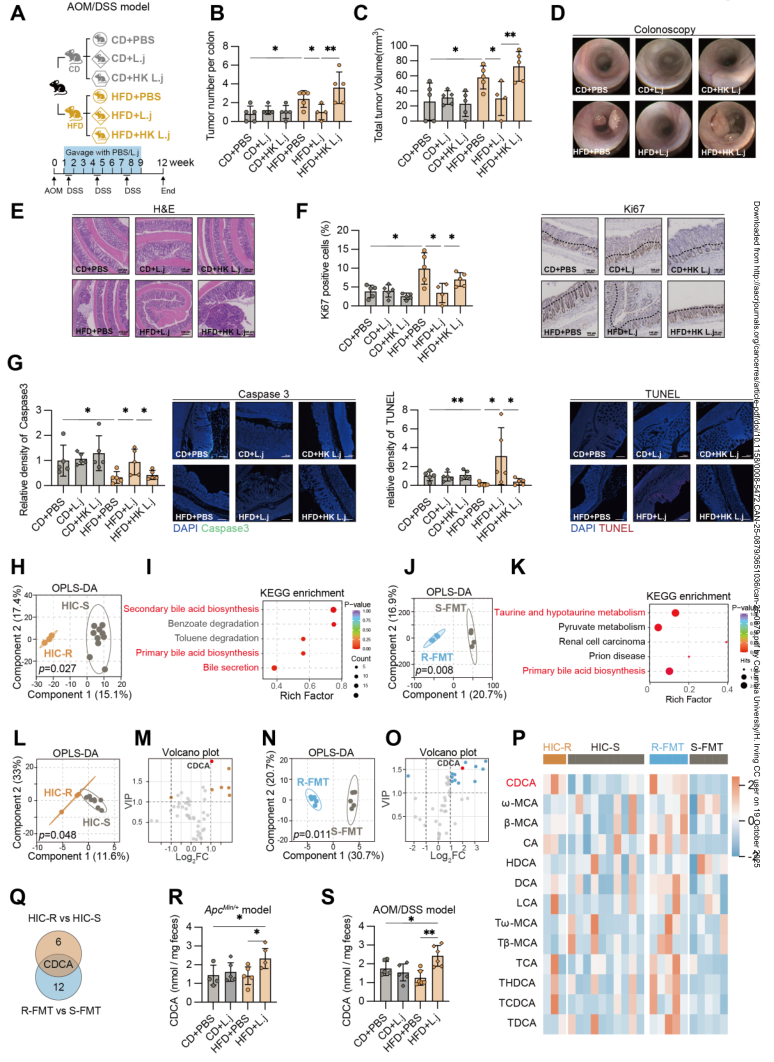
參考文獻:Liu C, Lai P, Hu J, et al. Lactobacillus johnsonii Synthesizes Chenodeoxycholic Acid to Reduce Susceptibility to High-Fat Diet-Induced Colorectal Cancer. Cancer Research. 2025
【陸】
Nature Metabolism | 適應膳食纖維的腸道菌群能夠清除膳食中的果糖并逆轉肝脂肪變性

過量攝入單糖果糖可導致肝臟脂肪生成過度以及腸道菌群失調,是引發心血管代謝疾病的一個風險因素。本研究發現,當腸道菌群適應膳食纖維菊粉時,會分解飲食中的果糖,并減輕或逆轉胰島素抵抗、肝臟脂肪變性和纖維化。菊粉還能激活肝臟的新生絲氨酸合成和胱氨酸攝取,增加谷胱甘肽的生成,保護肝臟免受果糖引起的脂質過氧化。這些分析為膳食纖維如何能夠促進腸道微生物群發揮作用、從而減輕宿主對有害營養物質的攝入以及減緩疾病進展提供了機制解釋。
1.肝臟脂質組學分析表明,補充菊粉減少了肝臟的脂質種類,包括鞘氨醇、鞘磷脂、二酰甘油和三酰甘油。qPCR分析表明,同時或延遲補充菊粉會下調由高果糖玉米糖漿誘導的肝臟纖維化標志物的基因表達。
2.通過氘代水(2H2O)示蹤探究了同步與延遲補充菊粉對肝臟DNL(新生脂肪生成)的影響,結果發現,小鼠單獨喂食高果糖玉米糖漿(HFCS) 14周后,2H標記的脂肪酸顯著增加,而同步或延遲補充菊粉均能抑制或逆轉這種誘導效應。
3.通過13C-果糖同位素示蹤技術分析發現,菊粉組小鼠肝臟線粒體中13C標記酰基肉堿水平升高。進一步結合代謝組學分析發現,喂食果糖和菊粉的實驗鼠盲腸中果糖衍生的短鏈脂肪酸的生成減少了,但在小腸內容物中乙酸與丁'酸生成顯著增加,提示菊粉可調節腸道不同部位的果糖代謝途徑。
4.通過給小鼠喂養13C-果糖并基于肝臟代謝組學分析發現,HFCS加菊粉飲食的小鼠肝臟中,由果糖合成的甘氨酸和絲氨酸比例顯著增加。
5.通過16S rRNA測序分析發現,菊粉補充使大腸中的產酸擬桿菌(B. acidifaciens)顯著富集,且產酸擬桿菌與肝臟DNL呈顯著負相關,與肝臟絲氨酸合成呈顯著正相關。
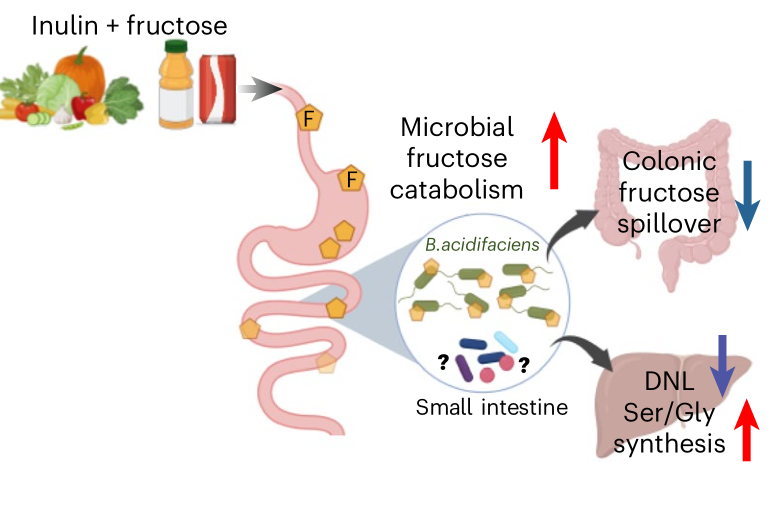
參考文獻:Jung S, Bae H, Song WS, et al. Dietary fibre-adapted gut microbiome clears dietary fructose and reverses hepatic steatosis. Nature Metabolism. 2025
【柒】
Gut | 果糖通過調控癌癥相關成纖維細胞與腫瘤細胞間的交互作用驅動結直腸癌進展

結直腸癌(CRC)發病率持續上升,腫瘤微環境(TME)中的癌癥相關成纖維細胞(CAFs)通過分泌細胞因子與代謝重編程,與癌細胞形成“共生回路”,腫瘤增殖、侵襲和耐藥。果糖作為膳食單糖,其轉運體GLUT5(SLC2A5)在多種癌細胞中異常高表達,可直接驅動腫瘤惡性表型,但果糖是否同樣被CAFs利用并介導腫瘤-基質互作仍空白。本研究整合473例CRC臨床標本、單細胞測序、代謝示蹤及多種基因工程小鼠模型,系統解析果糖在CAFs中的代謝命運及其對腫瘤-基質通訊的調控作用。
1.CAFs表達GLUT5,其水平與腫瘤浸潤深度、TNM分期及患者不良預后正相關;在多癌種中亦觀察到GLUT5+CAFs,提示該現象具有普遍性。
2.果糖可直接被CAFs攝取,經氧化磷酸化生成ATP,增強其增殖、遷移及激活標志物α-SMA/FSP-1表達;抑制GLUT5或果糖代謝酶KHK可逆轉上述效應。
3.腫瘤細胞利用果糖后釋放核苷酸及氨基酸(組氨酸、賴氨酸等),這些代謝物通過激活CAFs的Erk信號通路上調CXCL14表達;阻斷代謝物轉運或Erk磷酸化均可抑制CXCL14升高。
4.CXCL14+CAFs通過ACKR2/CXCR4受體反向激活腫瘤細胞的Erk通路,促進上皮-間質轉化EMT、遷移和遠處轉移;敲低CAFs中CXCL14或腫瘤細胞中其受體均顯著削弱果糖誘導的轉移能力。
5.小鼠模型顯示,高果糖飲食或GLUT5過表達加速原位瘤生長和肺轉移,而GLUT5敲除或CXCL14抑制可抵消果糖促瘤效應,證實果糖-CXCL14軸是治療潛在靶點。
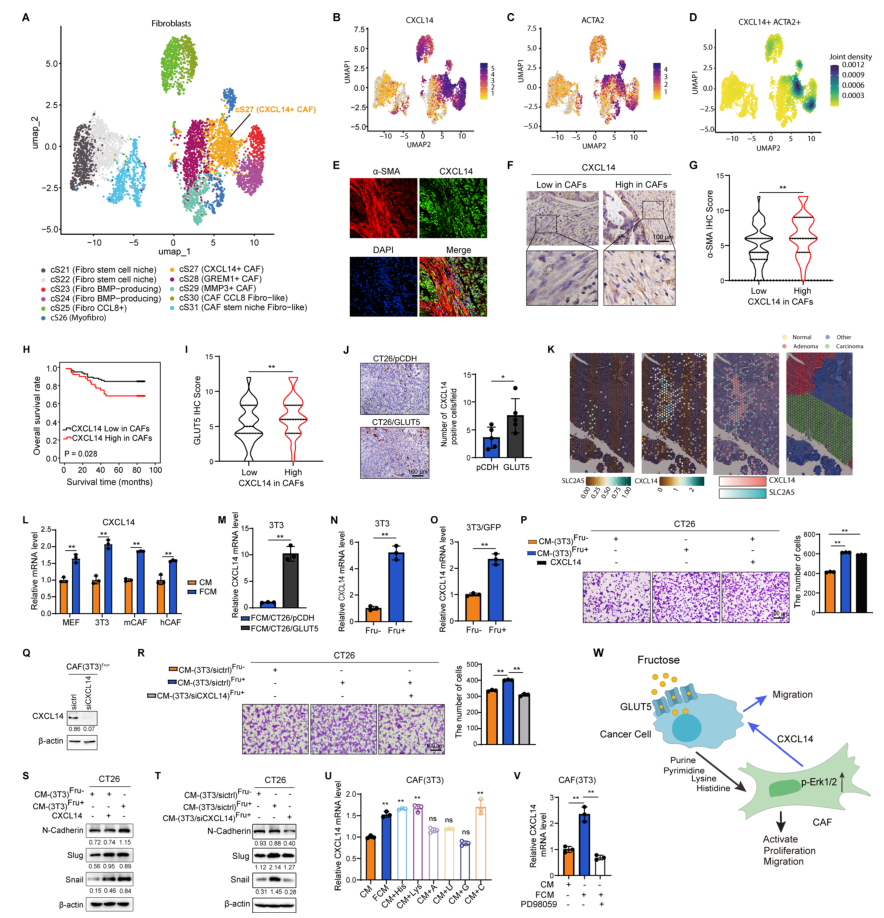
參考文獻:Cui, Y., Liu, H., Zhang, L., et al. Fructose drives colorectal cancer progression by regulating crosstalk between cancer-associated fibroblasts and tumour cells. Gut. 2025
【捌】
Nature | 杏仁核-肝臟信號通路協調應激下的血糖反應

在遭遇威脅時,生物體需在數分鐘內動員能量,表現為血糖升高和食欲抑制,傳統觀點將此歸因為下丘腦-垂體-腎上腺軸及腎上'腺素的快速分泌。然而,臨床研究發現慢性應激人群即使激素水平正常,仍易出現糖耐量受損和2型糖尿病,提示存在未被闡明的腦-肝直接通路。內側杏仁核(MeA)是情緒與威脅感知的核心,其下游密集投射至調控代謝的腹內側下丘腦(VMH),但MeA是否可獨立于經典內分泌軸、直接指揮肝臟糖異生仍未知。本研究結合化學遺傳、光遺傳、病毒跨突觸追蹤及13C示蹤技術,系統解析MeA-VMH-肝臟環路在急性與反復應激中的血糖調控作用。
1.各類急性應激(束縛、社交、電擊、視覺)均迅速激活MeA神經元,其鈣信號峰值早于血糖上升,提示MeA是應激性高血糖的“啟動器”。
2.特異性激活MeA→VMH谷氨酸或GABA神經元可在15分鐘內使血糖升高20–40mg/dL,同時抑制進食,而血漿皮質酮、胰島素、胰高血糖素和腎上'腺素水平無變化,證明該通路獨立于經典激素軸。
3.病毒示蹤顯示MeA經VMH→交感神經→腹腔神經節形成多突觸通路直抵肝臟;激活MeAVMH神經元顯著提高肝臟糖異生限速酶G6pc和轉錄因子FOXO1的表達,13C-丙酮酸示蹤證實肝臟M2-葡萄糖、M2-檸檬酸等中間產物增加30–50%,說明糖異生通量增強。
4.抑制或敲除MeA-VMH神經元可完全阻斷急性應激誘導的高血糖,卻不影響基礎血糖、糖耐量及應激激素水平,進一步確立該環路的“專職”應激血糖角色。
5.反復應激使MeA與MeAVMH神經元鈣反應逐次衰減,血糖升高幅度下降;慢性沉默或消融MeAVMH的小鼠在高脂飲食下表現出持續高血糖、糖耐量受損和肝G6pc上調,重現人類應激相關代謝異常的早期表型。
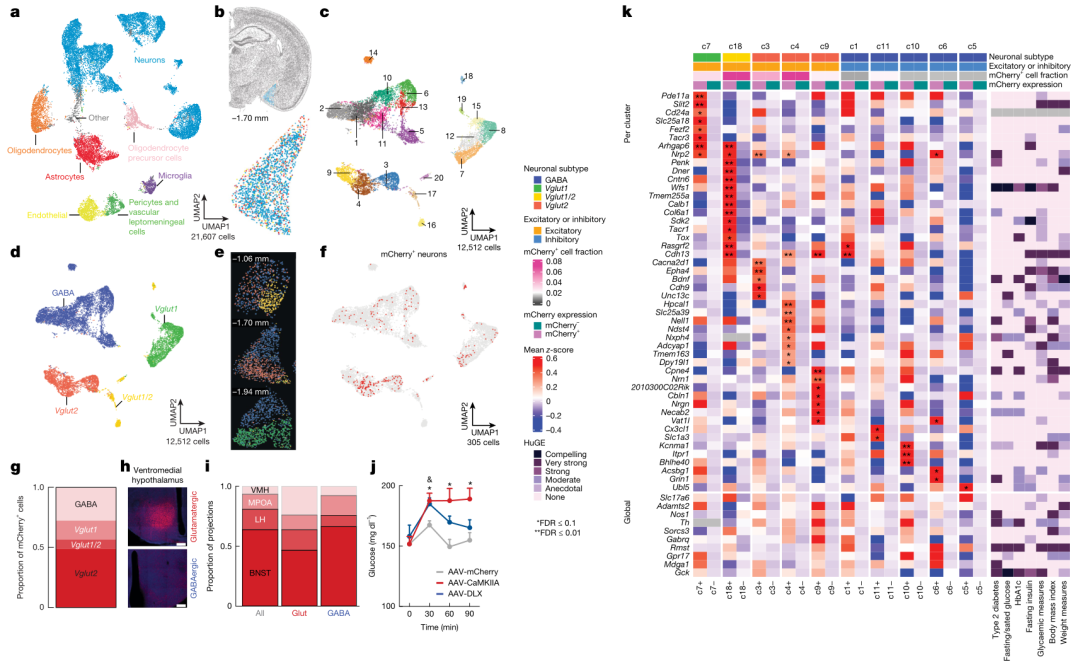
參考文獻:Carty, J. R. E., Devarakonda, K., O’Connor, R. M., et al. (2025). Amygdala–liver signaling orchestrates glycaemic responses to stress. Nature. 2025