巨噬細胞作為先天免疫系統的核心組成部分,能夠動態呈現多種功能狀態。例如,在感知到脂多糖(LBS)和干擾素-γ(IFNγ)等與感染相關的經典激活信號后,巨噬細胞首先啟動促炎功能以清除病原體;隨后這些反應逐漸減弱,并轉變為促進組織修復與炎癥消退的表型。這一功能轉換常伴隨代謝的動態重編程,后者在巨噬細胞的免疫應答中起關鍵作用。
近十年來,越來越多研究致力于識別不同功能狀態下的巨噬細胞代謝特征,并解析關鍵代謝變化如何調控其免疫功能。理解免疫應答中代謝重編程的機制與意義具有廣泛重要性,然而,除中心碳代謝和脂代謝途徑以外,巨噬細胞在更廣泛代謝網絡中的特性仍待深入探索。
2025年8月,美國威斯康星州莫格里奇研究所的Jing Fan教授團隊在Nature Metabolism上發表了題為“Classically activated macrophages undergo functionally significant nucleotide metabolism remodelling driven by nitric oxide”的研究結果,揭示了巨噬細胞在經典激活過程中核苷酸代謝的全面重編程,發現一氧化氮(NO)是這一過程的關鍵調控因子,并闡明了這種代謝重編程的功能意義。

一. 技術路線圖

二. 研究結果
1.刺激后,巨噬細胞的核苷酸代謝發生重編程
首先,通過對巨噬細胞M1極化過程的代謝組學分析,研究者發現LPS+IFNγ刺激24小時后,核苷酸代謝(包括嘌呤、嘧啶及磷酸戊糖途徑)與精氨酸代謝、TCA循環等共同發生顯著重編程。持續或急性刺激均引發核苷酸代謝明顯改變,表現為IMP積累和dTTP耗竭,且持續刺激效應更強,而急性刺激下的變化在72-96小時可逆,提示該過程具有動態適應性。
轉錄組分析進一步支持上述發現:基因表達呈時間依賴性變化,并在96小時恢復近基線狀態。核苷酸降解相關基因(如Pnp1、Pnp2、Upp1)早期顯著上調,而合成基因(如Rrm1、Rrm2)則下調。GO分析中“核苷堿基化合物代謝過程調控”條目顯著富集,從轉錄層面確認了核苷酸代謝在M1激活中的關鍵重編程作用。
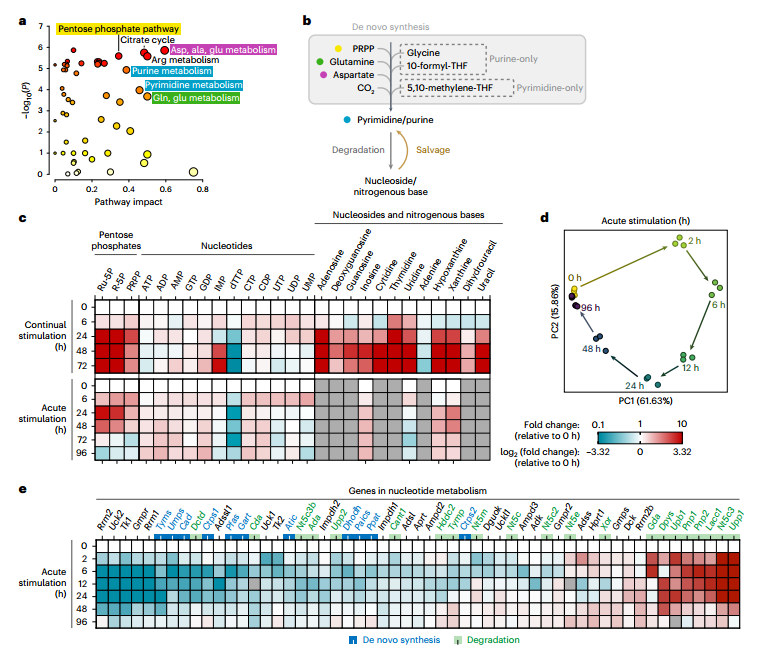
圖1. 多組學分析發現核苷酸代謝重編程
2.核苷酸由從頭合成轉向補救合成
為解析巨噬細胞經典激活中核苷酸代謝通量的變化,研究者采用穩定同位素示蹤技術進行了多維度分析。γ-¹?N-谷氨酰胺標記實驗顯示,盡管刺激后UMP的標記比例上升,但其下游產物dTMP、CTP和dCTP的標記完全消失,提示由三磷酸胞苷合成酶(CTPS)和胸苷酸合成酶(TYMS)催化的嘧啶核苷酸合成途徑被顯著阻斷。同樣的,在嘌呤合成通路中,關鍵中間體AICAR仍可被標記,但其下游產物IMP的標記比例大幅下降,表明AICAR向IMP的轉化過程受阻。
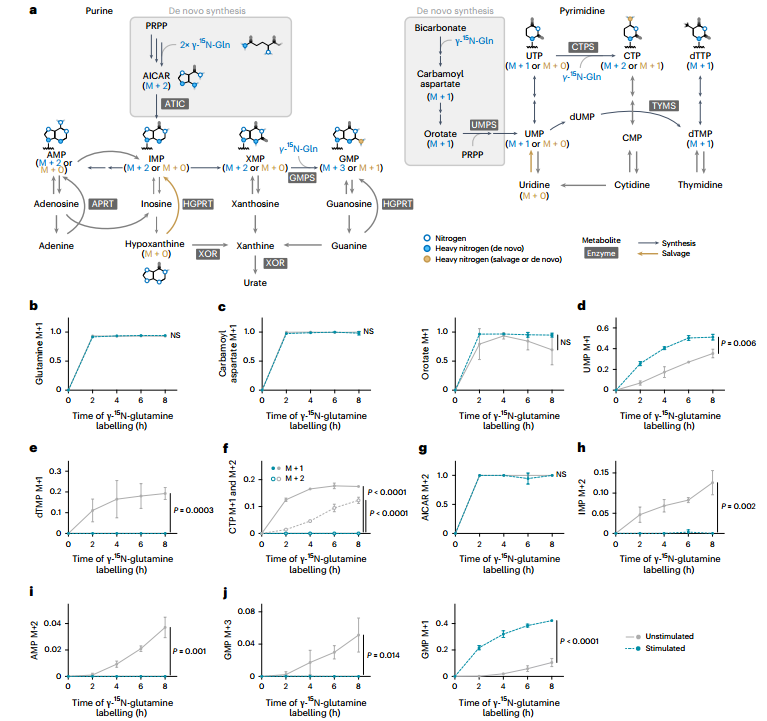
圖2. 谷氨酰胺代謝流顯示核苷酸從頭合成阻滯
值得注意的是,盡管從頭合成被抑制,胞內多數嘌呤核苷酸水平仍維持穩定,暗示存在替代途徑進行補給。進一步通過¹?N標記模式區分合成途徑發現:刺激后,含3個標記氮(M+3)的GMP(從頭合成來源)幾乎消失,而含1個標記氮(M+1)的GMP(補救合成來源)顯著增加,證實細胞轉向依賴補救合成以維持嘌呤核苷酸穩態。U-¹³C-葡萄糖標記實驗也支持補救途徑的通量增強。
為直接驗證外源嘌呤的利用能力,研究者使用U-¹?N-肌苷進行示蹤,發現刺激后細胞對肌苷的攝取和轉化為IMP的效率顯著提高。此外,在Hprt1(次黃嘌呤-鳥嘌呤磷酸核糖轉移酶,參與嘌呤補救合成)敲除細胞中,刺激導致的IMP積累現象幾乎完全消失,證明HGPRT介導的堿基補救途徑是維持IMP水平的關鍵機制。
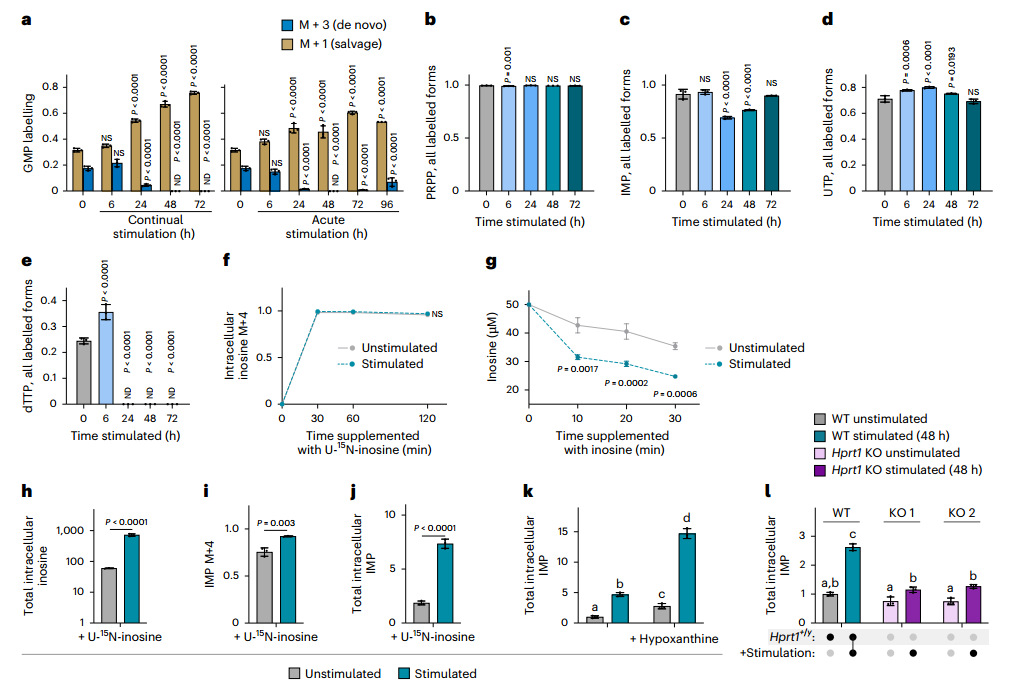
圖3. 巨噬細胞轉為嘌呤補救合成
3.核苷酸降解增強,但嘌呤堿基的完全氧化被阻斷
先前的代謝組學顯示,刺激后最顯著變化之一是核苷和含氮堿基積累,這些核苷酸降解產物可作為補救合成底物。RNA-seq進一步證實,關鍵核苷酸降解酶(如PNP1、PNP2、UPP1、LACC1)表達顯著上調,提示核苷酸降解發生重編程。為驗證這點,研究者通過U-¹³C-葡萄糖的代謝流檢測發現,刺激后BMDMs中IMP降解為肌苷的速率顯著加快,與肌苷積累現象一致。此外,刺激不僅增加胞內核苷/堿基水平,還促進其向培養基釋放,表明核苷降解為堿基的過程也得到增強。
隨后,U-¹?N-肌苷代謝流直接檢測了核苷酸的降解能力,結果顯示刺激后巨噬細胞將肌苷轉化為次黃嘌呤的速率提升,而其經黃嘌呤氧化還原酶(XOR)向黃嘌呤和尿酸的轉化卻受到顯著抑制。這種代謝流轉向使次黃嘌呤得以通過HGPRT補救為IMP。
為進一步驗證PNP在次黃嘌呤積累和為補救合成提供底物中的作用,研究者用PNP抑制劑福達拉濱處理巨噬細胞。如預期所示,PNP抑制劑福達拉濱處理完全阻斷刺激誘導的次黃嘌呤和IMP積累,并降低補救合成來源的GMP比例。這與Hprt1敲除結果共同證實:刺激通過增強PNP活性促進次黃嘌呤生成,同時抑制XOR介導的完全降解,使代謝流轉向HGPRT依賴的補救合成,最終驅動IMP積累。
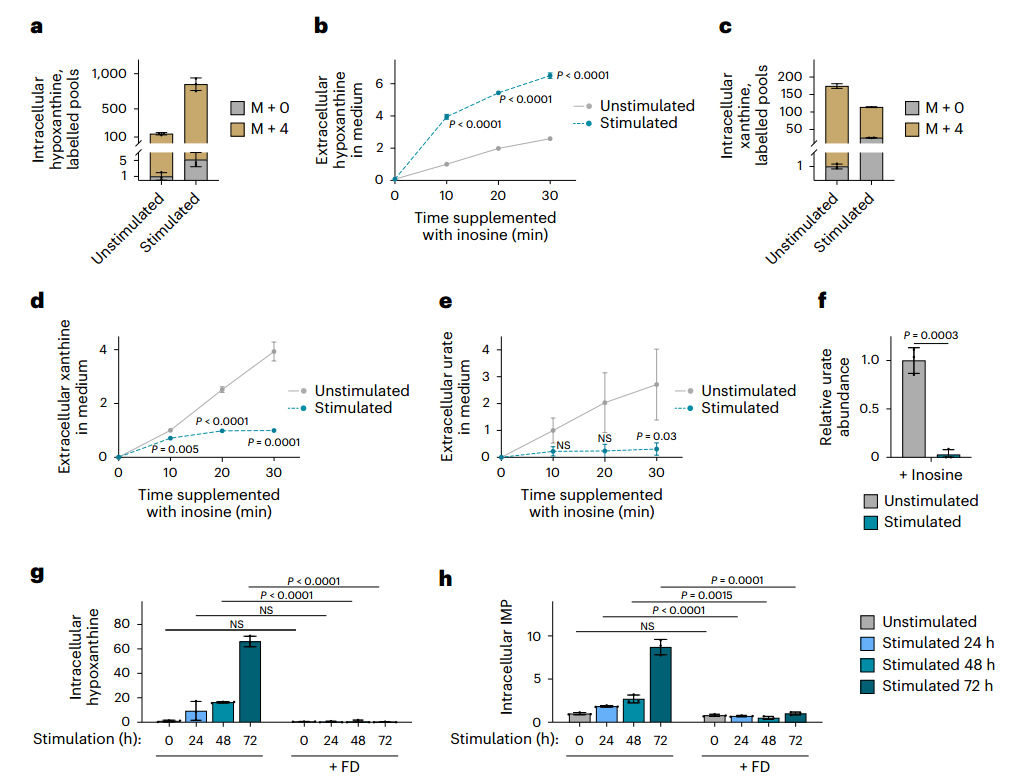
圖4. 受刺激后,巨噬細胞的核苷酸降解發生變化
4.NO是核苷酸代謝重編程的關鍵驅動因子
關于核苷酸代謝重編程的分子機制,研究者首先提出了兩種主流假設并逐一進行驗證。第一種假設認為,核苷酸從頭合成抑制可能源于一碳單位供應不足。然而,外源性補充一碳供體(如甲酸)及四氫葉酸輔因子(亞葉酸)均未能恢復核苷酸的合成,表明一碳代謝缺陷并非主要原因。第二種假說則聚焦于能量還原應激或前體物質匱乏,即NAD?/NADH比率失衡或天冬氨酸短缺。研究者在補充丙酮酸后,雖成功逆轉了NAD?/NADH的下降并恢復了天冬氨酸水平,但核苷酸的從頭合成仍處于抑制狀態,從而排除了這一可能性。
在否定上述常見機制后,研究者轉而從時序關聯中尋找線索。通過動態監測發現,核苷酸合成抑制的發生時間與誘導型一氧化氮合酶(iNOS,由Nos2編碼)的激活及一氧化氮(NO)大量產生高度同步。Western Blot及代謝組學數據證實,iNOS蛋白上調及其催化產物瓜氨酸的積累早于IMP和dTTP合成的受阻。轉錄組分析進一步顯示,在Nos2誘導之后,胸苷酸合成酶(TYMS)編碼基因Tyms等核苷酸合成關鍵基因的表達顯著下調,提示NO可能調控這些基因的轉錄,進而關閉合成通路。
為確立NO的因果地位,研究者進行了功能增益與損失實驗:在Nos2敲除(Nos2?/?)的巨噬細胞中,炎癥刺激引發的核苷酸合成抑制被顯著挽救;而在未刺激細胞中外源添加NO供體,則足以模擬出與炎癥刺激類似的核苷酸抑制表型。綜上所述,研究者通過嚴謹的排他性驗證和時序?功能分析,確立了NO是巨噬細胞在炎癥應激下核苷酸代謝重編程的關鍵驅動因子。
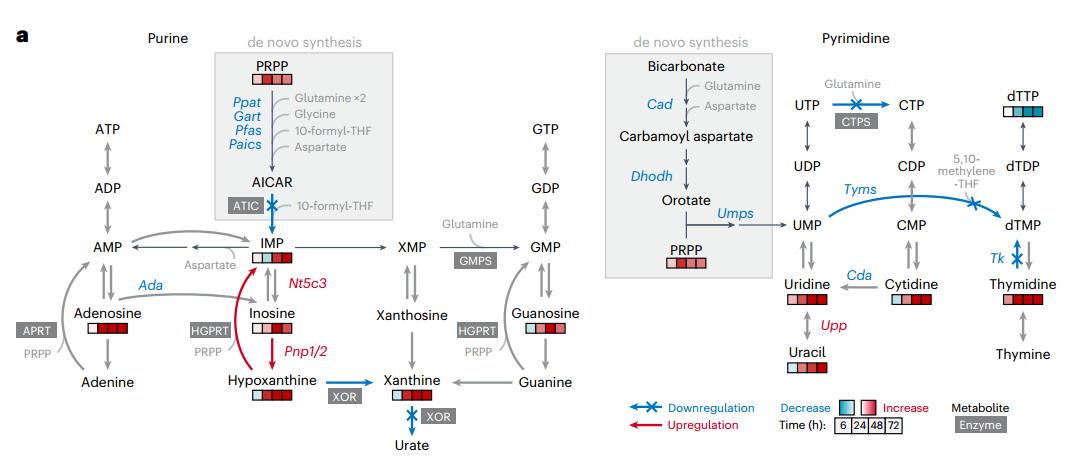
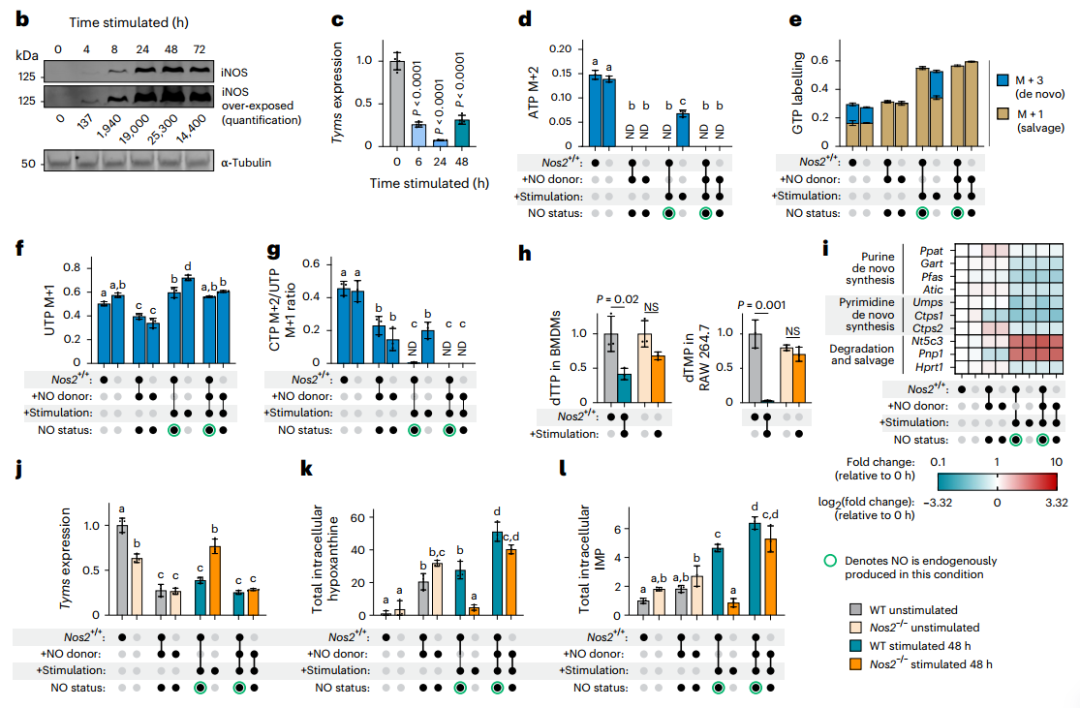
圖5. NO在核苷酸代謝重編程中起關鍵作用
5.巨噬細胞通過核苷酸代謝重編程增強免疫防御
最后,研究者探討了這種代謝重編程的功能意義。結果表明,由HGPRT介導的嘌呤補救合成對巨噬細胞的免疫功能具有關鍵作用。通過基因敲除(Hprt1 KO)或藥物抑制(6-MP)阻斷該途徑后,巨噬細胞多項免疫功能受損:包括免疫應答基因(如 Ptgs2、Mmp14)表達下降、IL-10等細胞因子分泌減少、遷移能力減弱,以及對細菌顆粒的吞噬效率顯著降低。更重要的是,在弓形蟲感染模型中,研究發現宿主細胞的嘌呤補救合成能力直接限制病原體的復制。在Hprt1 KO的巨噬細胞中,由于無法有效競爭嘌呤底物,弓形蟲增殖速度顯著加快。
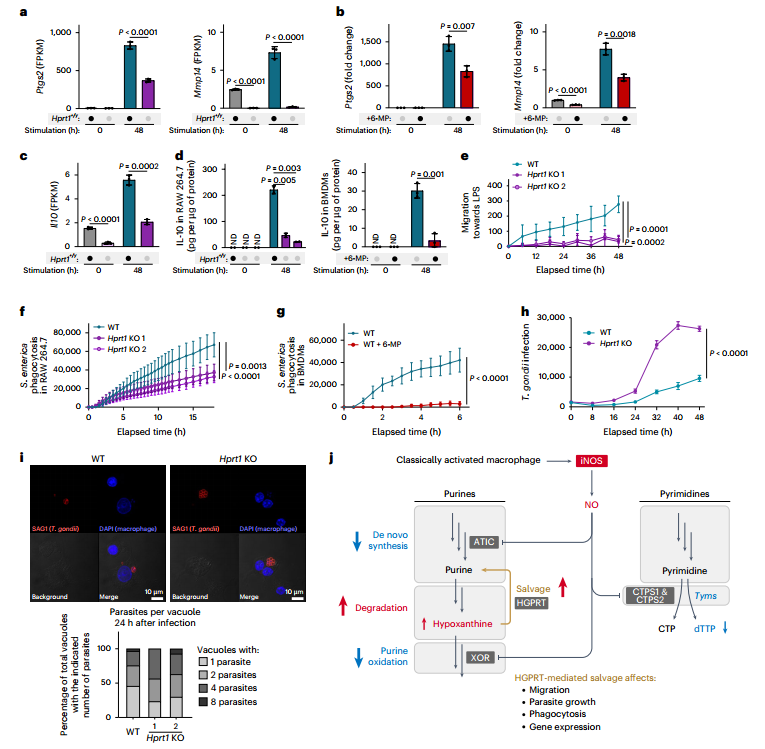
圖6. 核苷酸代謝的改變影響巨噬細胞功能,抑制病原體增殖
綜上所述,巨噬細胞在激活后轉向補救合成不僅是一種維持自身核苷酸水平的代謝適應,更是一種主動的免疫策略:一方面支撐其遷移、吞噬和細胞因子產生等效應功能;另一方面通過在代謝層面競爭嘌呤資源,直接抑制細胞內病原體的增殖。
三、全文總結
本研究通過代謝組和代謝流檢測發現,核苷酸代謝是巨噬細胞經典激活過程中變化最為顯著的代謝通路之一。具體而言,嘌呤和嘧啶的從頭合成被抑制,核苷酸降解為含氮堿基的過程被上調,但嘌呤堿基的完全氧化受到抑制,從而使代謝轉向補救合成。從機制上,一氧化氮被確定為核苷酸代謝的關鍵調控因子,它同時驅動多個關鍵變化;干擾嘌呤補救合成后,可改變許多刺激應答相關基因的表達、抑制巨噬細胞的遷移與吞噬功能,并促進胞內寄生蟲的增殖。
總之,本研究系統揭示了巨噬細胞經典激活過程中核苷酸代謝的動態重編程,并闡明了其調控機制與功能意義。
參考文獻
John SV, Seim GL, Erazo-Flores BJ, et al. Classically activated macrophages undergo functionally significant nucleotide metabolism remodelling driven by nitric oxide. Nat Metab. 2025.
繪譜幫你測
本文利用代謝組學和多層級代謝流分析逐層闡釋一氧化氮為核苷酸代謝的關鍵調控因子。麥特繪譜提供全面的靶向、非靶向代謝組學和代謝流檢測服務,可追蹤含13C和15N等被標記物100+種,全面覆蓋糖酵解和TCA循環通路、磷酸戊糖途徑、 氨基酸代謝、脂肪酸代謝、 一碳代謝、 核苷酸代謝通路等。豐富的個性化標記定制經驗--[U-13C6]-Fructose,[U-13C16]-Palmitate, [U-13C3]-Serine,[U-13C2]-Glycine, [U-13C3]-Alanine, [U-13C3]-Pyruvate, [U-13C4]Malic Acid, [U-13C18]-Oleic Acid, 13CO2, 15N-NH4CL, [1, 2-13C2]-Glucose, [2,3,3-D3]-Serine, [2,3-13C2]Alanine, [1,2,3-13C3]-Choline等。歷經數年項目積累,檢測各類貼壁細胞、懸浮細胞、菌體、培養液、線粒體、組織、糞便等樣本類型,涵蓋多發性骨髓瘤、肝癌、線粒體遺傳代謝病、免疫細胞活性與疾病、心血管疾病等多個研究方向,合作項目成果突出,文章平均IF>10+。
麥特繪譜開創性地搭建了醫學領域高端代謝組學技術平臺,覆蓋了非靶向-全定量-代謝流等全方位的高端醫學代謝組解決方案,同時全面布局微生物組學、轉錄組學和蛋白質組學等多組學技術服務,已成為全球多組學研究者的優選合作伙伴。麥特繪譜已為數百家三甲醫院、科研院所和企業提供多組學一站式整體解決方案,協助客戶與合作伙伴發表SCI文章600+篇,累計影響因子6000+,平均IF>10,涵蓋Cell, Science, Nature, Cancer Cell, Signal Trans-duction and Targeted Therapy, Nature Biotechnology, Cell Metabolism等權威期刊。