研究背景
支鏈氨基酸在心臟生理學和疾病中發揮著關鍵作用,纈氨酸分解代謝酶ACAD8的遺傳缺陷在臨床上與人類異丁酰輔酶A失調和心肌病相關,但其在心臟病中的作用仍不清楚。近期中國醫學科學院北京協和醫學院劉德培院士、陳厚早教授團隊聯合四川大學華西第二醫院唐小強教授團隊在Nature Communications(IF 15.7)上發表文章“ACAD8 deficiency promotes pathological cardiac hypertrophy in response to pressure overload by regulating histone isobutyrylation”,闡明了ACAD8缺陷在心臟病中的作用,并揭示了轉錄調控和心臟病理學中的組蛋白異丁酰化。
· 維真助力 - AAV·
基因信息
ACAD8:酰基輔酶A脫氫酶家族成員8
實驗動物
9周齡雄性C57BL/6小鼠
病毒產品
AAV9-cTnT-ACAD8、AAV9-cTnT-Ctrl
注射方式
尾靜脈注射
注射量
0.8×1012 vg,100μl of viral dilution
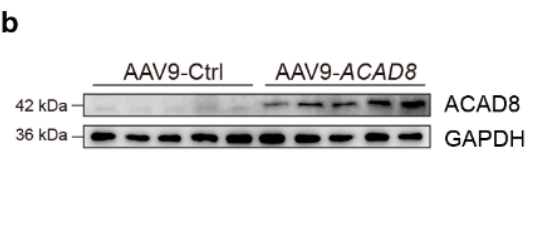
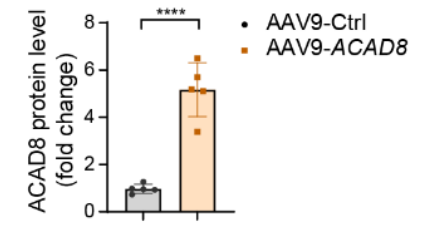
WB實驗表明AAV9介導的ACAD8在小鼠體內成功過表達
結果展示
1. 心肌細胞特異性Acad8敲除會加劇小鼠壓力超負荷時的心臟肥大
作者發現病理性心臟肥大中發生纈氨酸分解代謝受損和ACAD8缺乏,表明其可能在心臟肥大中發揮關鍵作用。為了研究纈氨酸分解代謝和ACAD8在病理性心臟肥大中的作用,作者構建了心肌細胞特異性Acad8敲除小鼠(Acad8cKO),并通過TAC手術誘導小鼠病理性心臟肥大。發現與對照小鼠相比,Acad8cKO小鼠表現出心臟功能障礙加劇,此外心臟重量與體重(HW/BW)和心臟重量與脛骨長度(HW/TL)的比率增加,同時心臟的橫截面積和心肌細胞增大,心臟纖維化增加,以及肥大標記基因(Nppa和Nppb)表達增加。心臟肥大和心力衰竭進展常常伴隨著細胞死亡,作者還使用TUNEL測定評估了心臟組織的細胞凋亡,發現Acad8cKO小鼠表現出更嚴重的細胞死亡。這些發現表明ACAD8缺陷直接促進病理性心臟肥大。
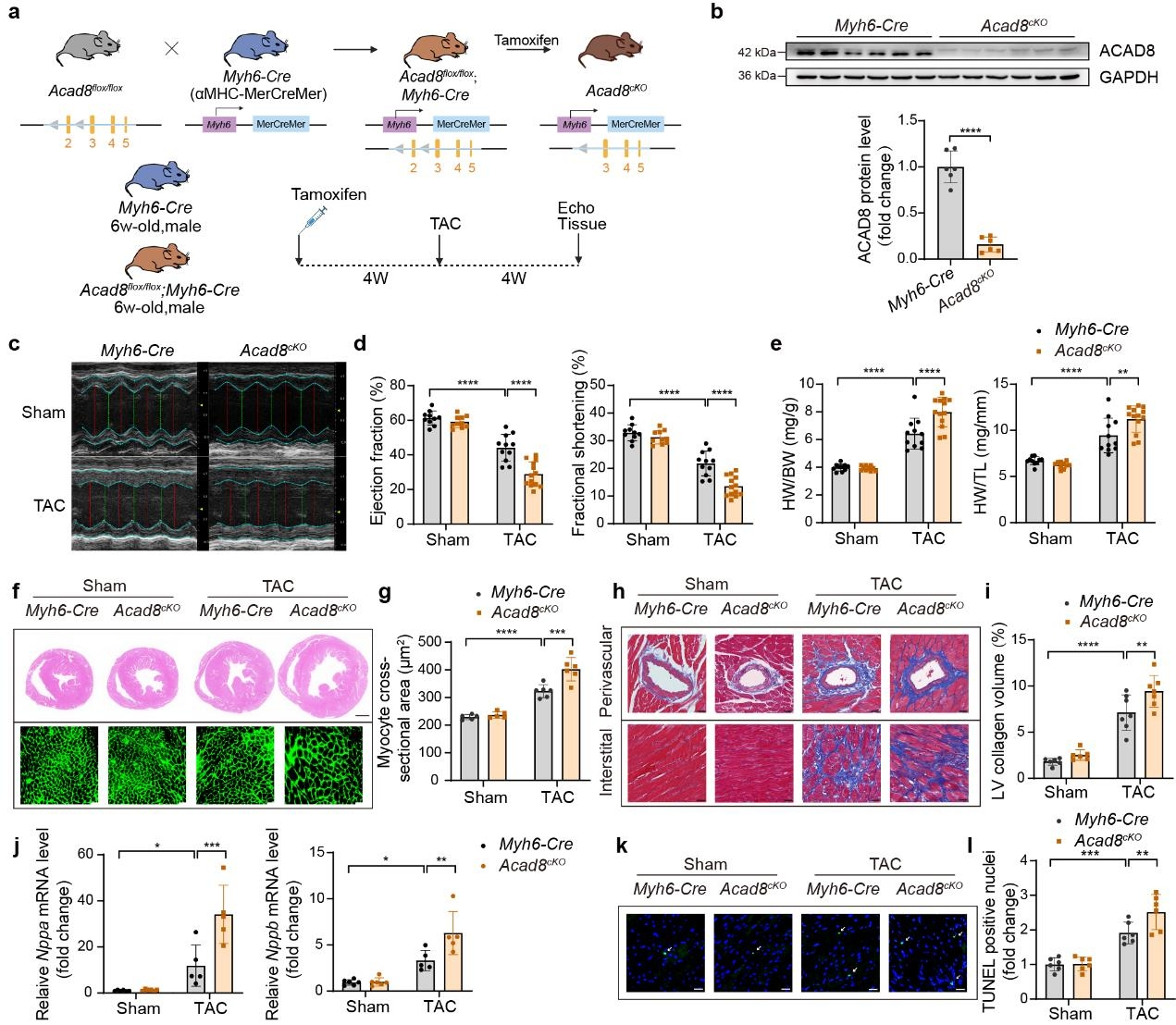
圖1 心肌細胞特異性Acad8敲除加劇TAC誘導的病理性心臟肥大
2. ACAD8缺陷引起的異丁酰輔酶A積累通過調節染色質可及性和TEAD2富集促進心肌細胞肥大
作者發現TAC手術心臟中ACAD8缺陷導致異丁酰輔酶A顯著積累并促進組蛋白異丁酰化。對isobutyrate(異丁酰輔酶A供體)處理的NRCM(新生大鼠心肌細胞)和接受TAC的Acad8cKO小鼠心臟進行RNA測序分析,發現心肌病相關基因和心臟肥大標志物的上調;TRANSFAC和JASPAR PWM分析表明TEAD2是兩個RNA測序數據集中上調基因的轉錄因子。作者進一步在Nppa和Nppb的啟動子區域內鑒定了潛在的TEAD2結合基序,并發現isobutyrate處理和Acad8敲低均顯著增強了Nppa和Nppb啟動子區域中的TEAD2結合。此外TEAD2過表達促進Acad8敲低和isobutyrate治療誘導的Nppa表達。基于這些發現,作者推測異丁酰輔酶A和異丁酰化修飾的增加可能會促進染色質開放,從而增強肥大基因啟動子處的TEAD2結合。因此作者評估了染色質可及性的變化,發現isobutyrate處理和Acad8敲低均導致Nppa和Nppb啟動子區域染色質可及性增加。此外isobutyrate處理和Acad8敲低都促進了H3K9ibu和H3K23ibu在肥大相關基因Nppa啟動子區域的富集。這些結果表明,Acad8缺陷通過異丁酰輔酶A的積累來調節組蛋白異丁酰化、染色質可及性和目標基因啟動子區域TEAD2富集,從而導致心肌細胞肥大。
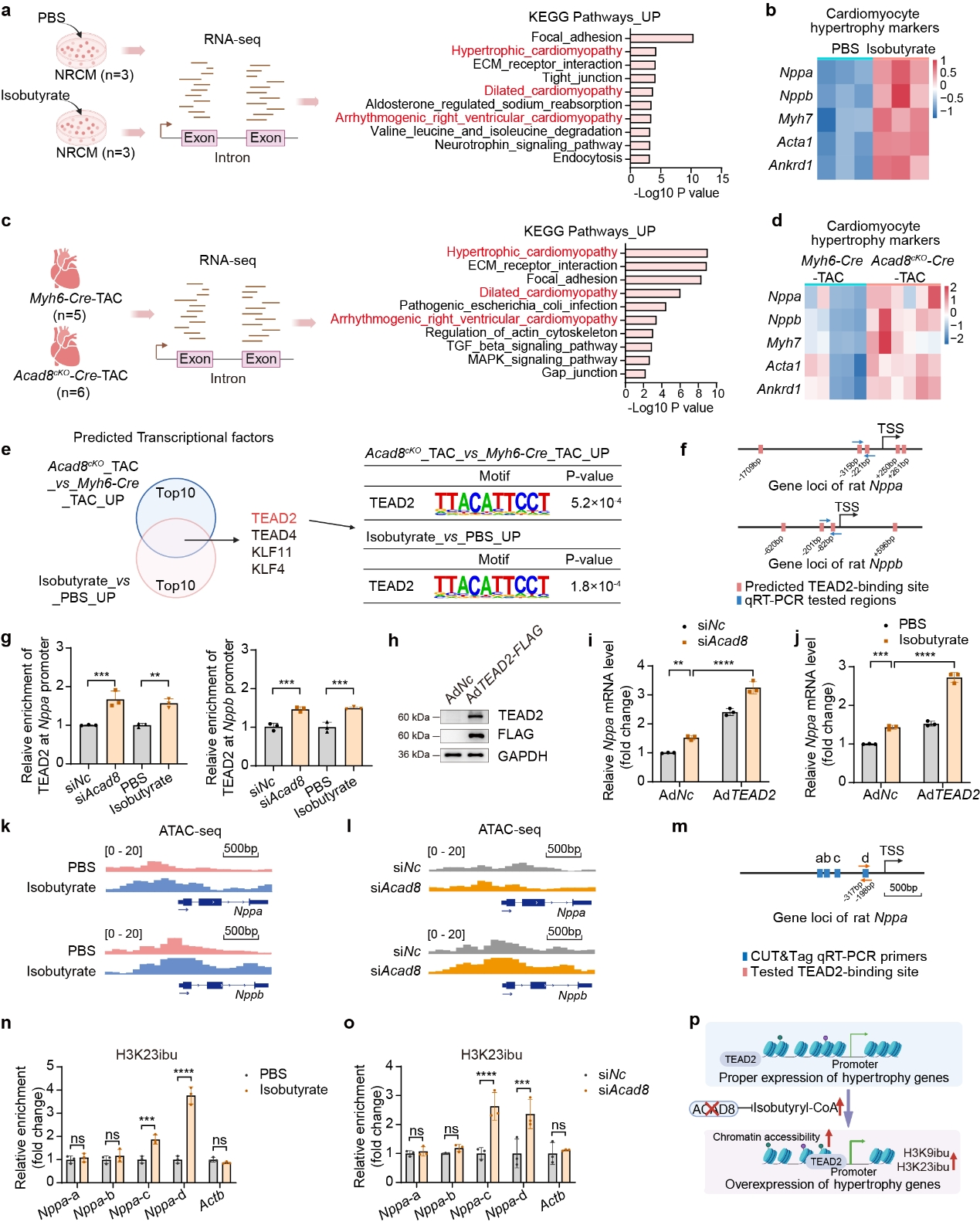
圖2 Acad8缺陷和isobutyrate通過調節染色質可及性和TEAD2富集促進肥大基因的表達
3. ACAD8過表達減弱TAC誘導的心臟肥大
作者建立了TAC誘導的病理性心臟肥大的小鼠模型,并在TAC 7天后通過靜脈注射AAV9-cTnT-Ctrl或AAV9-cTnT-ACAD8。結果顯示心肌細胞特異性ACAD8過表達有效抑制TAC誘導的心臟功能障礙,減輕了心臟重量,減少了心臟肥大和纖維化以及肥大相關基因表達水平,減弱了TAC誘導的細胞死亡。這些發現表明心肌細胞特異性ACAD8過表達抑制TAC誘導的心臟肥大并恢復心臟功能。機制研究發現ACAD8過表達通過降低異丁酰輔酶A水平和組蛋白異丁酰化來改善病理性心臟肥大。
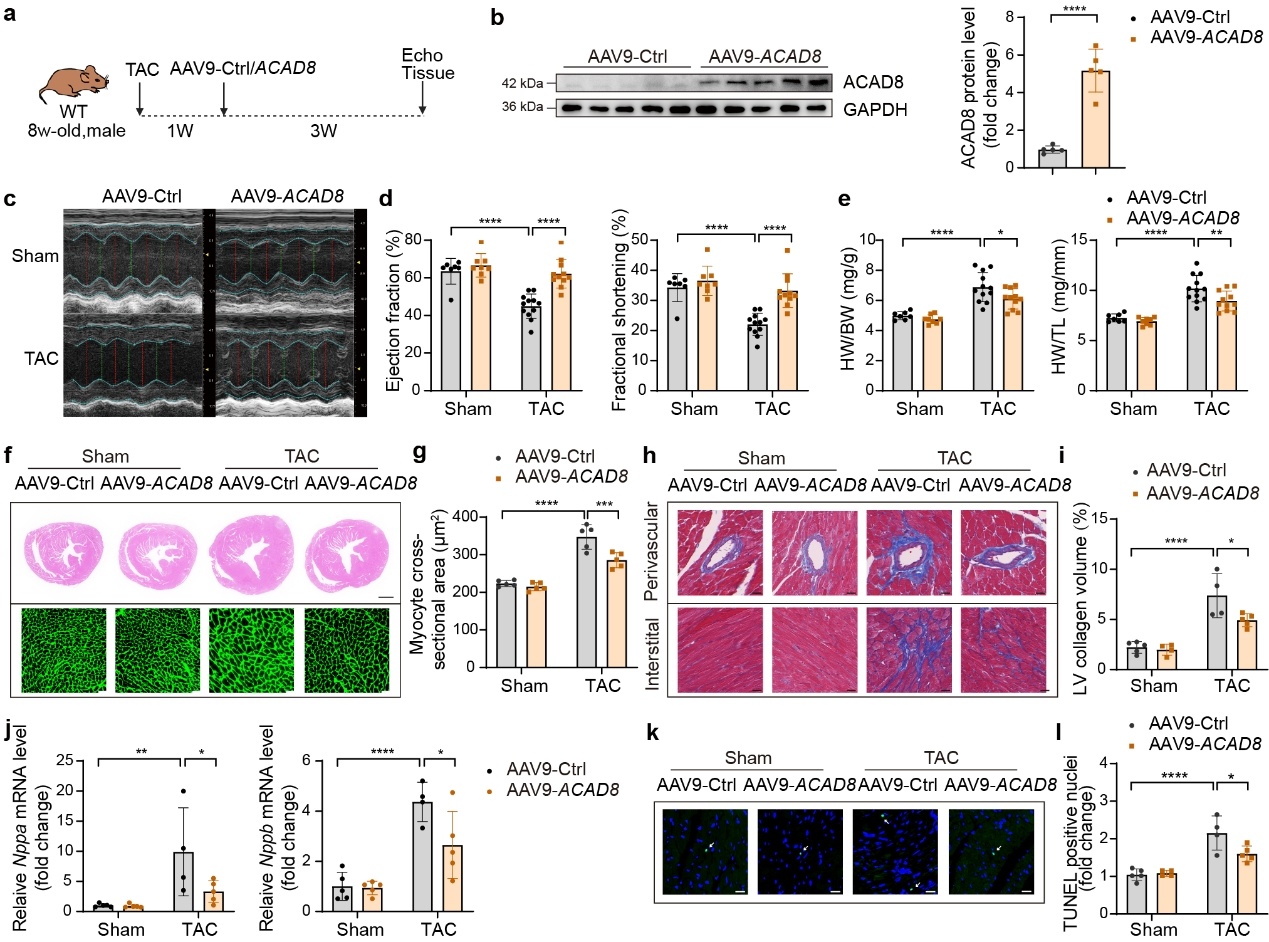
圖3 心肌細胞中AAV9介導ACAD8過表達可改善TAC誘導的病理性心臟肥大
實驗結論
本研究強調了ACAD8在調節異丁酰輔酶A水平以維持心臟穩態中的作用,Acad8缺陷誘導的異丁酰輔酶A積累通過表觀遺傳重編程加劇了壓力超負荷誘導的病理性心臟肥大。ACAD8-異丁酰輔酶A-異丁酰化軸可能是心臟肥大的新治療靶點。