
 期刊:Cancer Research
期刊:Cancer Research
影響因子:16.6
發表時間:2026年3月
通訊作者:南京醫科大學附屬腫瘤醫院尹榮、劉桐言、譚婧
伯豪生物技術服務:單細胞RNA測序服務
科學問題
EGFR突變型肺腺癌(LUAD)在接受酪氨酸激酶抑制劑(TKI,如奧希替尼,Osimertinib)治療時,腫瘤細胞如何逃避藥物殺傷進入“藥物耐受持久性(DTP)”狀態?微環境中的特定腫瘤相關成纖維細胞(CAF)如何通過與癌細胞形成物理連接,接收受損細胞器,從而協助癌細胞清除氧化應激并促進腫瘤耐藥及復發?
實驗材料和方法
患者隊列與樣本處理:
樣本分組:治療初治的EGFR突變型LUAD患者(n=15),以及奧希替尼耐藥復發組織與初治組織對比。
樣本類型:手術切除的新鮮組織(用于單細胞測序、原代細胞分離、類器官培養)。
細胞模型:患者來源的類器官(PDO)、原代分離的CAF亞群(RGS5+MYL9+CAFs等)、EGFR突變細胞系(HCC827, PC9)。
多組學數據與功能驗證:
單細胞轉錄組(scRNA-seq):解析EGFR突變LUAD中的成纖維細胞異質性。
轉錄組測序(Bulk RNA-seq):分析藥物耐受persister (DTP)細胞在不同共培養條件下的基因表達差異。
功能實驗:
三維類器官共培養藥敏測試、線粒體轉移示蹤(MitoTracker, mitoDsRed)、透射電鏡(TEM)、CRISPR/Cas9或shRNA敲低(Miro1, RhoA)、小鼠皮下移植瘤模型及藥物干預。
導語
南京醫科大學附屬腫瘤醫院尹榮、劉桐言、譚婧研究團隊在 EGFR 突變肺腺癌耐藥機制研究中取得重要突破。研究團隊借助伯豪生物提供的單細胞轉錄組測序技術支持,首次在EGFR突變型肺腺癌(LUAD)中鑒定出一類特殊的癌癥相關成纖維細胞亞群(RGS5+MYL9+CAFs)。研究發現,這些細胞在TKI藥物(如奧希替尼)誘導的壓力下,通過隧道納米管(TNTs)接收來自癌細胞轉移的受損線粒體,充當“代謝垃圾桶”,幫助癌細胞清除毒性活性氧(ROS),從而進入休眠耐受狀態(DTP)并引發腫瘤復發。該研究不僅揭示了腫瘤細胞與微環境間“代謝共生”的新機制,還證明了聯合使用EGFR-TKI與Rho激酶抑制劑(法舒地爾,Fasudil)可有效阻斷這一保護路徑,顯著克服耐藥并延緩腫瘤復發,為攻克EGFR-TKI耐藥提供了全新靶點與可行方案。
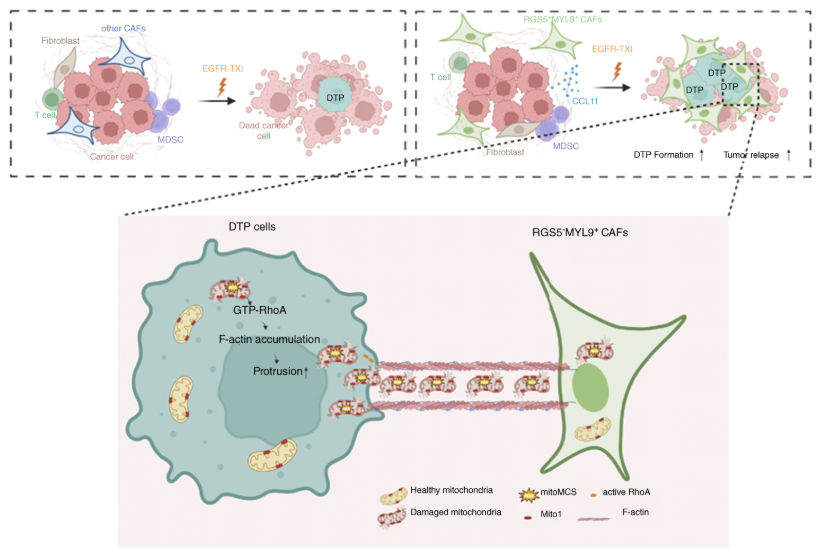
研究結果的示意概述
技術服務
單細胞RNA測序服務
(技術服務伯豪生物提供)
主要研究
1. 單細胞圖譜揭示耐藥相關的CAF新亞群
通過對6例未接受過治療的EGFR突變LUAD患者樣本進行單細胞測序,研究團隊繪制了精細的成纖維細胞圖譜,鑒定出5個CAF亞群。其中,類器官-CAF共培養顯示,RGS5+MYL9+CAFs可顯著降低奧希替尼殺傷效果,促進停藥后腫瘤快速復發。臨床樣本分析顯示,該亞群在耐藥復發組織中的浸潤顯著增加,且在TCGA數據庫中高浸潤該亞群的患者生存期更短。
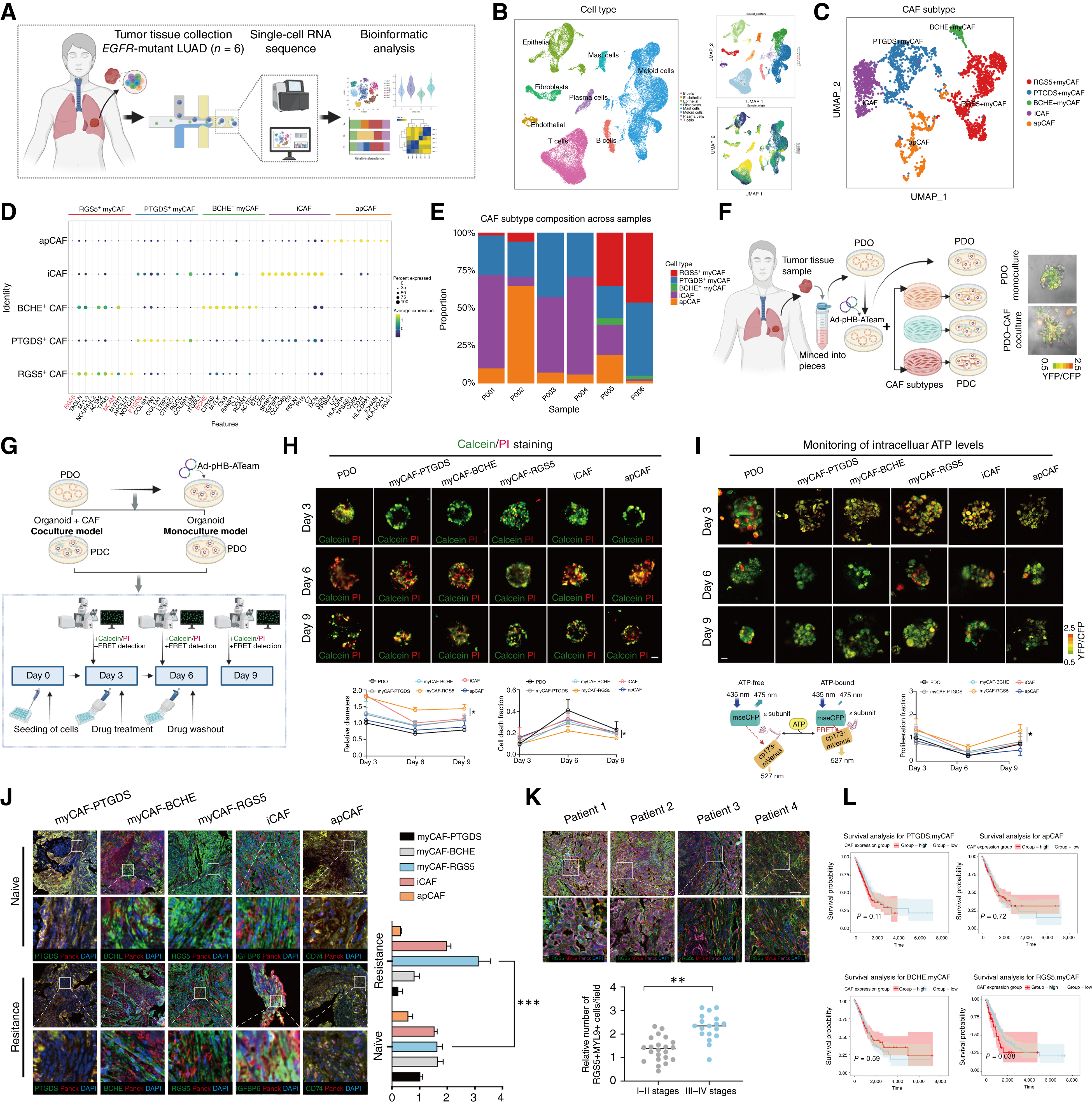
圖1. RGS5+MYL9+CAFs與EGFR突變LUAD的EGFR-TKI抗性相關
2. RGS5+MYL9+CAFs充當“代謝垃圾桶”,清除癌細胞損傷線粒體
腫瘤細胞是如何在奧希替尼的高壓下存活的?研究發現,RGS5+MYL9+CAFs的共培養顯著降低了TKI處理下腫瘤細胞的線粒體ROS(mtROS)水平,維持其線粒體膜電位。但RGS5+MYL9+CAFs自身的mtROS水平反而激增,出現線粒體功能障礙。體內小鼠模型同樣證實,這群CAF通過“接收”癌細胞的損傷線粒體,幫助腫瘤細胞解毒并維持存活。
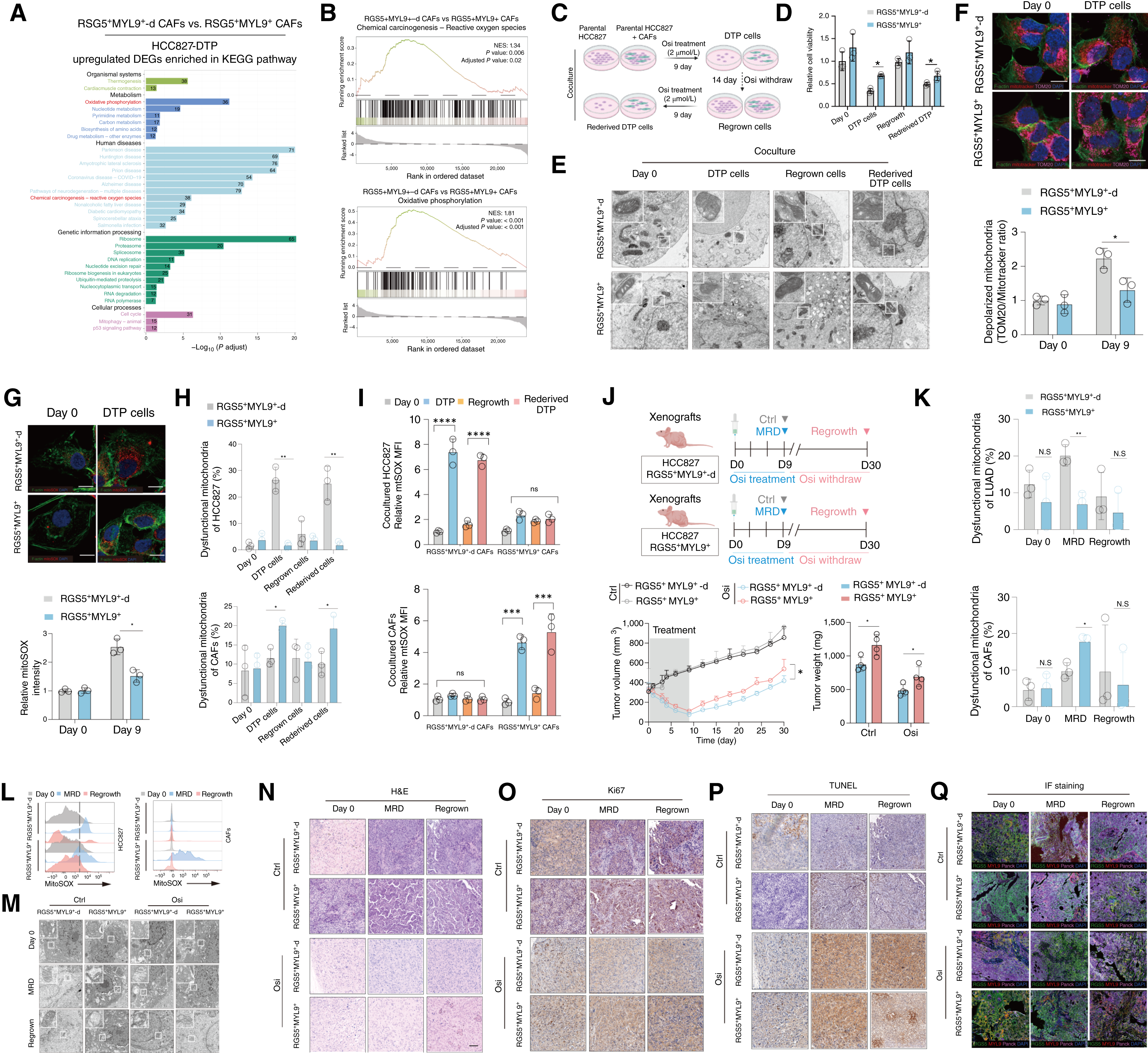
圖2. RGS5+MYL9+CAFs可緩解EGFR-TKI誘導的線粒體損傷,并促進DTP形成
3. 隧道納米管(TNT)是線粒體單向轉運的核心通道
受損線粒體如何實現跨細胞移動?借助共聚焦顯微鏡與流式細胞術追蹤,研究團隊發現,在奧希替尼的處理下,耐藥的腫瘤細胞能夠生長出由F-肌動蛋白(F-actin)支撐的細長細胞膜突起——隧道納米管(TNTs),并與RGS5+MYL9+CAFs精準連接。受損(去極化)的線粒體正是沿著該“通道”被輸送給CAF。使用細胞松弛素D(阻斷肌動蛋白聚合)可直接切斷納米管,完全阻斷線粒體的轉移,從而恢復腫瘤對TKI的敏感性。
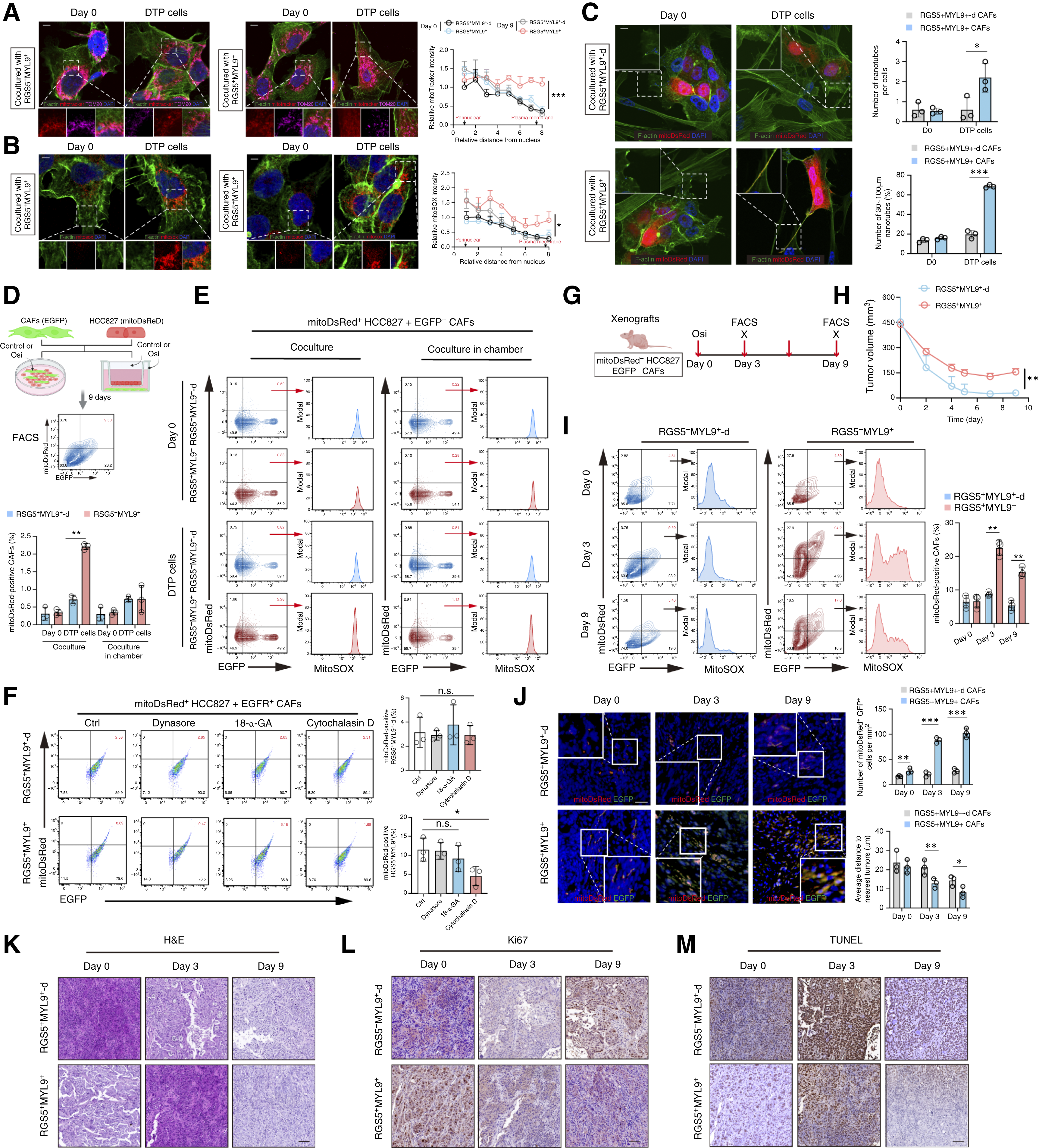
圖3. 細胞間納米管使受損線粒體從LUAD細胞轉運到RGS5+MYL9+CAFs成為可能
4. mtROS通過激活Miro1和RhoA驅動納米管的形成
機制探究表明,TKI藥物引發的初始mtROS為信號源,上調腫瘤細胞中線粒體Rho GTP酶1(Miro1)的表達,并驅動受損線粒體從核周向細胞膜周緣聚集。同時,mtROS激活關鍵細胞骨架調節蛋白RhoA。活躍的GTP-RhoA促進了F-肌動蛋白在細胞膜上的局部富集,進而拉伸形成納米管。敲低Miro1或利用抗氧化劑清除mtROS,均可阻斷線粒體轉運、逆轉腫瘤耐藥性。
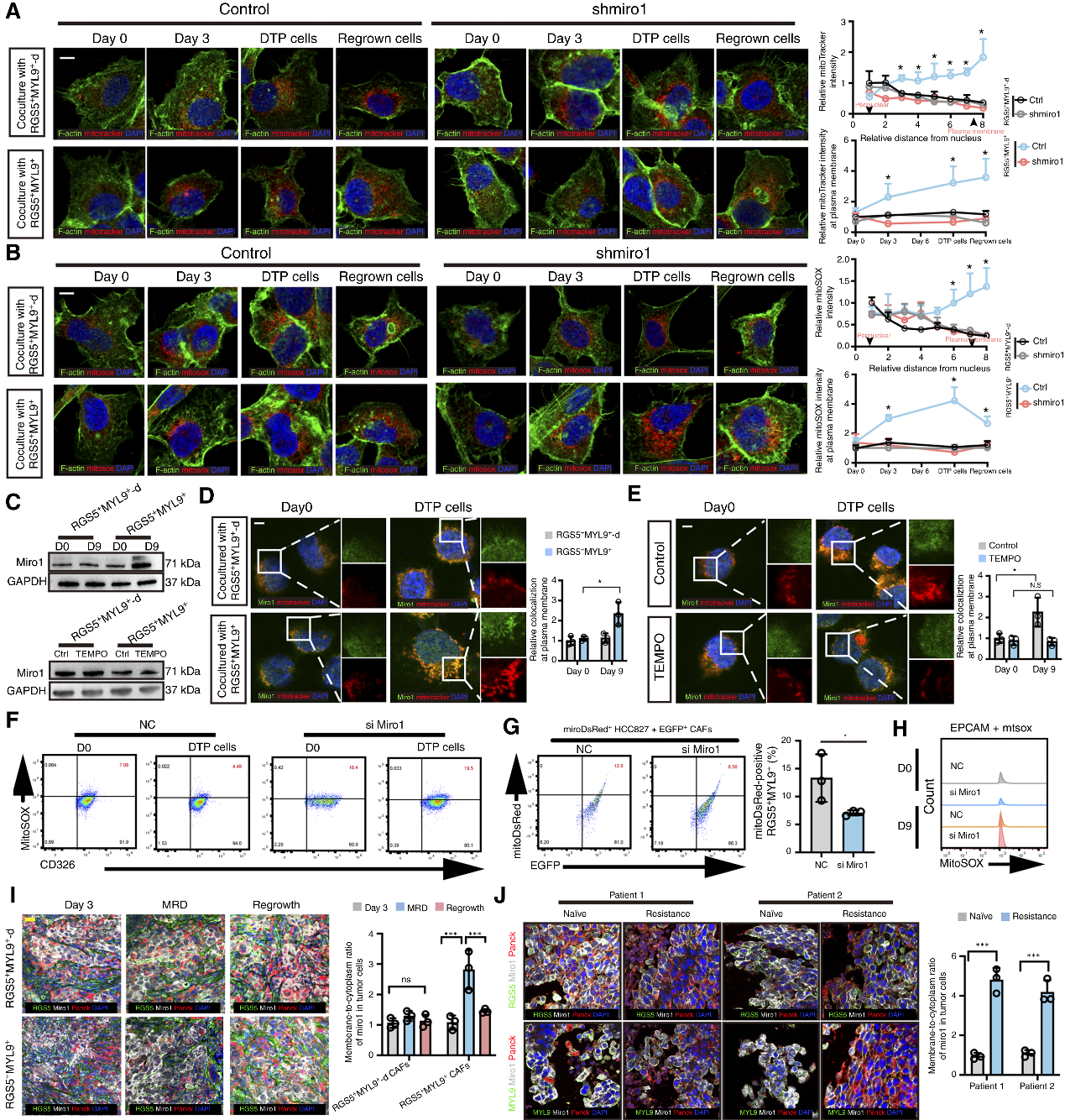
圖4. mtROS上調Miro1以調節DTP細胞受損線粒體的分布
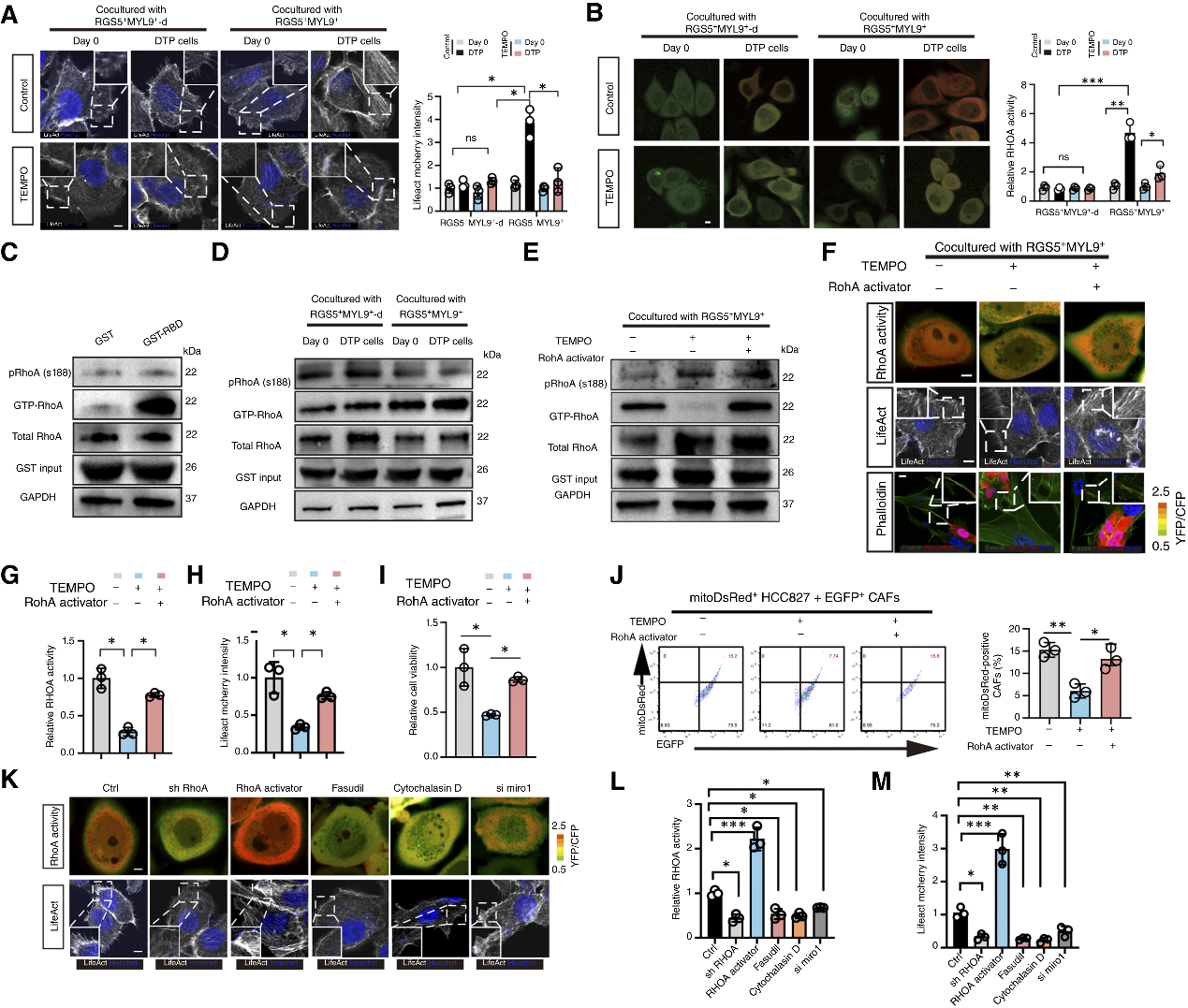
圖5. 依賴RhoA的F-肌動蛋白積累介導納米管的形成和線粒體轉移
上述過程共同發揮作用,便可降低癌細胞內的氧化應激,使其轉化為藥物耐受的慢循環狀態(DTP),而接受線粒體的CAF自身則出現線粒體功能受損。
5. 趨化招募:DTP細胞通過CCL11“招兵買馬”
ELISA結果顯示,處于DTP狀態的癌細胞會分泌趨化因子CCL11,特異性招募RGS5+MYL9+CAFs向腫瘤部位遷移(形成極近的空間距離以便于納米管對接)。空間定位顯示:CAF與殘留腫瘤細胞緊密接觸,形成“耐藥保護微環境”。這種“主動招募”機制使得CAF能夠及時到達并為癌細胞提供代謝支持。在小鼠模型中,使用CCL11中和抗體可阻斷CAF的浸潤,顯著恢復腫瘤對奧希替尼的敏感性并延緩復發。
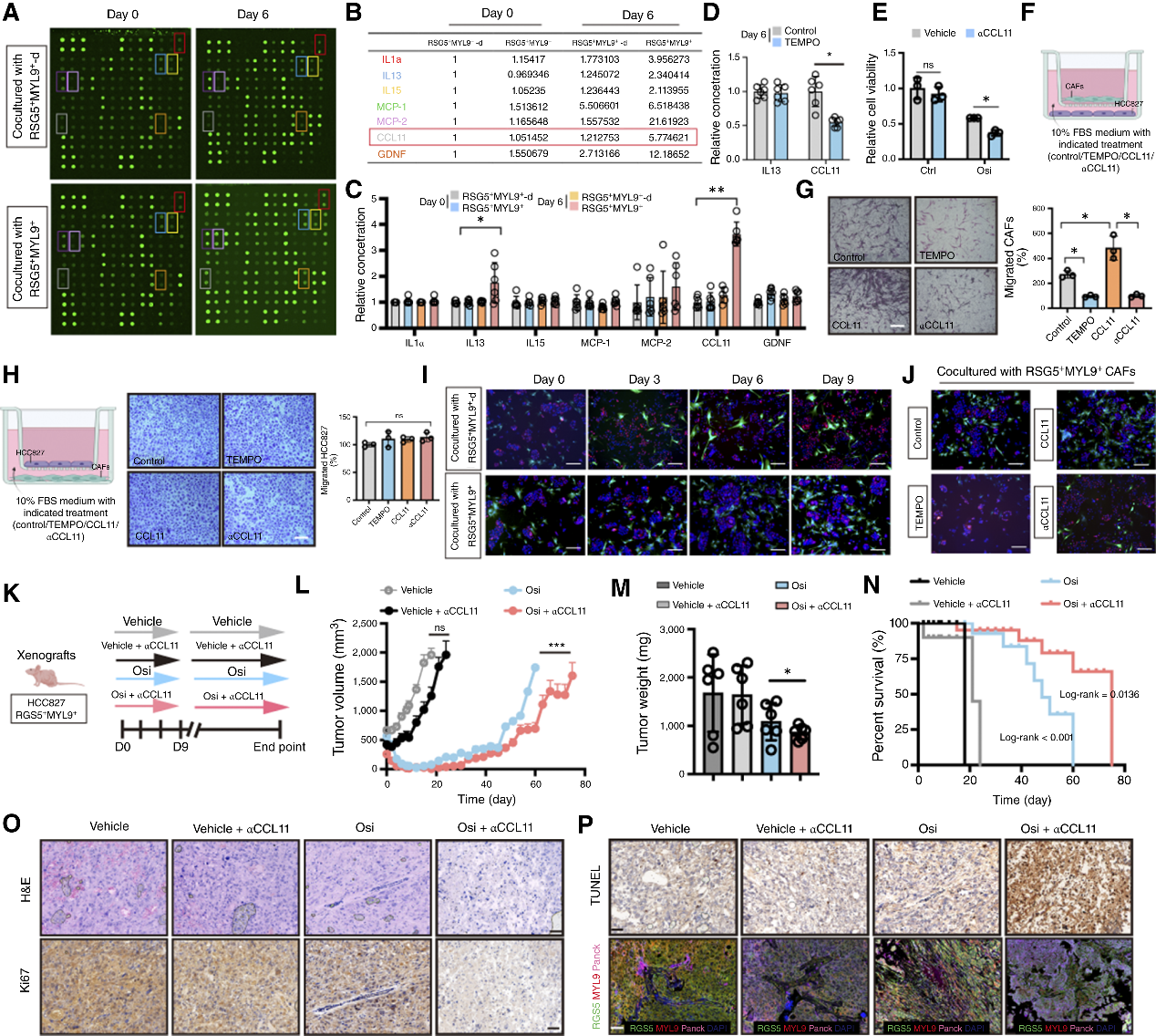
圖6. DTP細胞通過CCL11分泌招募RGS5+MYL9+CAFs
6. 治療策略:法舒地爾聯合奧希替尼克服耐藥、延緩腫瘤復發
基于上述機制,研究團隊探索了聯合治療策略。研究人員在動物模型中測試了FDA已批準的Rho激酶抑制劑法舒地爾(Fasudil,已被證明能有效抑制RhoA介導的TNTs形成)與奧希替尼的聯合用藥。實驗結果表明:在人源化小鼠模型中,“奧希替尼+法舒地爾”的聯合方案相比單藥治療,顯著減少了腫瘤內的CAF互作,增加了癌細胞凋亡,并大幅延長小鼠的生存期。
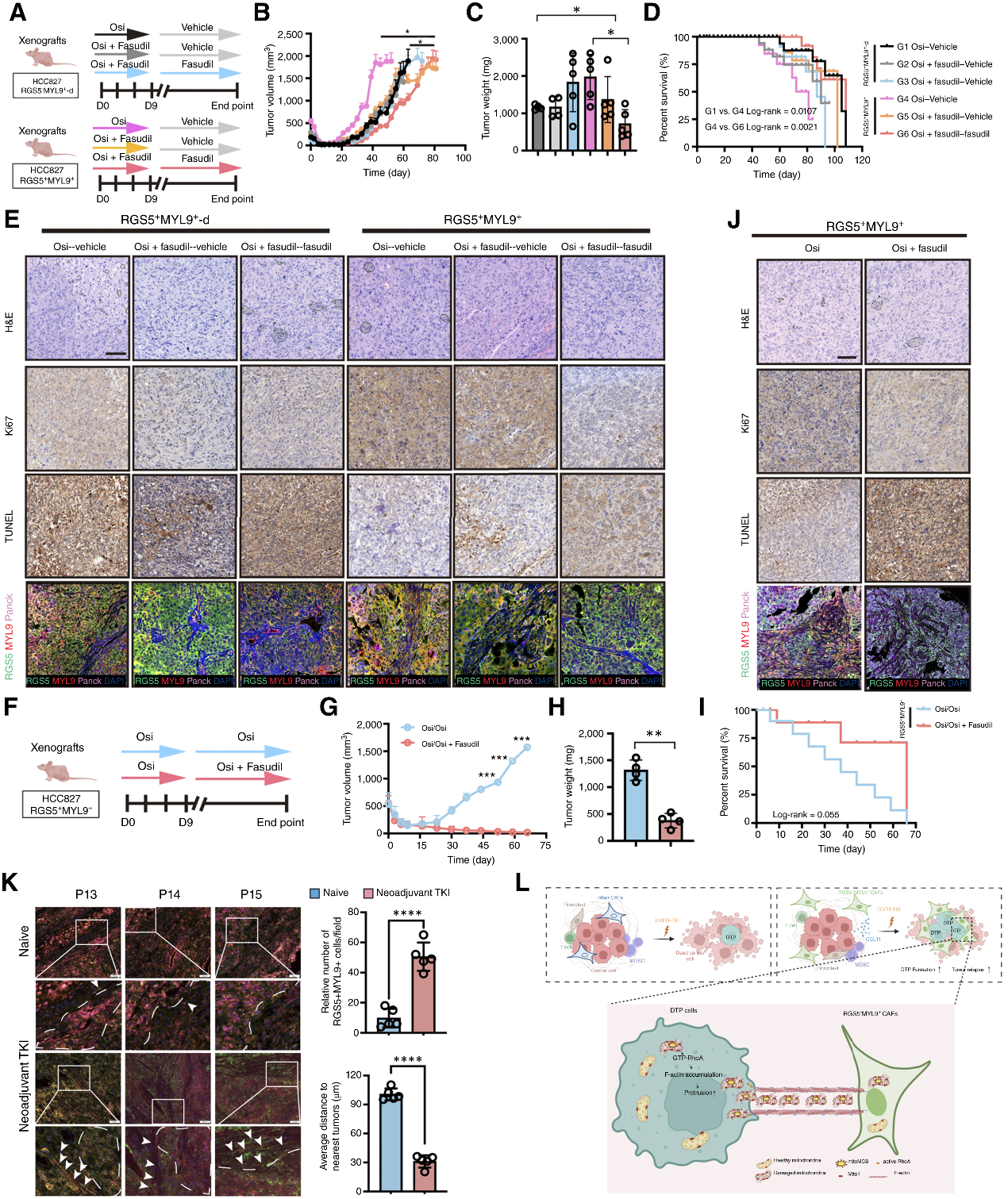
圖7. 使用法舒地爾和奧希替尼聯合靶向治療納米管,從而抑制持久耐藥細胞(DTP)的形成
結論
本研究借助單細胞轉錄組系統性解析 EGFR 突變肺腺癌 CAF 異質性,首次闡明RGS5+MYL9+ CAF 通過納米管介導的損傷線粒體轉運幫助癌細胞清除氧化應激、形成 DTP 耐藥狀態的全新機制。研究鎖定CCL11、Miro1、RhoA為關鍵干預靶點,并提出奧希替尼 + 法舒地爾這一極具臨床轉化潛力的組合療法,為臨床克服 EGFR-TKI 耐藥、減少腫瘤復發提供了全新機制與治療策略。
參考文獻:
Liu, Tongyan et al. “Transfer of Damaged Mitochondria from Cancer Cells to Cancer-Associated Fibroblasts Promotes Tyrosine Kinase Inhibitor Tolerance in EGFR-Mutant Lung Cancer.” Cancer research vol. 86,5 (2026): 1215-1231. doi:10.1158/0008-5472.CAN-25-0433