在癌癥進展和免疫干預過程中,腫瘤特異性T細胞持續暴露于抗原刺激,逐步陷入功能耗竭狀態(TEX)。這一過程并非驟然發生,而是經歷精確的階段性分化:最初是具有自我更新潛能的祖細胞狀態(TEXprog),隨后退化為功能受限的效應樣狀態(TEXeff),最終淪為徹底失能的終末耗竭狀態(TEXterm)。在這三種細胞亞群中,TEXprog是免疫治療的最后希望,此階段T細胞對免疫檢查點阻斷治療(ICB)仍具敏感性,而進入TEXterm后則難以逆轉。
已有機制研究表明,T細胞耗竭的本質是代謝重編程與表觀遺傳調控失衡的協同作用。以組蛋白乙酰化修飾為例:在TEX細胞中,代謝重編程引起核內乙酰輔酶A區域性耗竭,導致H3K27ac等關鍵乙酰化標記在抗腫瘤基因(如IFN-γ、TNF-α)啟動子區顯著減少,形成轉錄沉默狀態。這種耗竭表型通過DNA甲基化等機制穩定遺傳,形成自我強化的惡性循環。
因此,研究者們思考:若能用某些方法矯正TEXprog的代謝-表觀失衡,或許能阻止它發展成終末狀態,這未嘗不是一種免疫治療的新思路。2024年12月,美國索爾克生物研究所的Susan M. Kaech團隊在Science發表了題為“Nutrient-driven histone code determines exhausted CD8+T cell fates”的研究論文,聚焦TEX細胞代謝與表觀遺傳的互作機制,為優化腫瘤治療策略提供了潛在干預靶點。

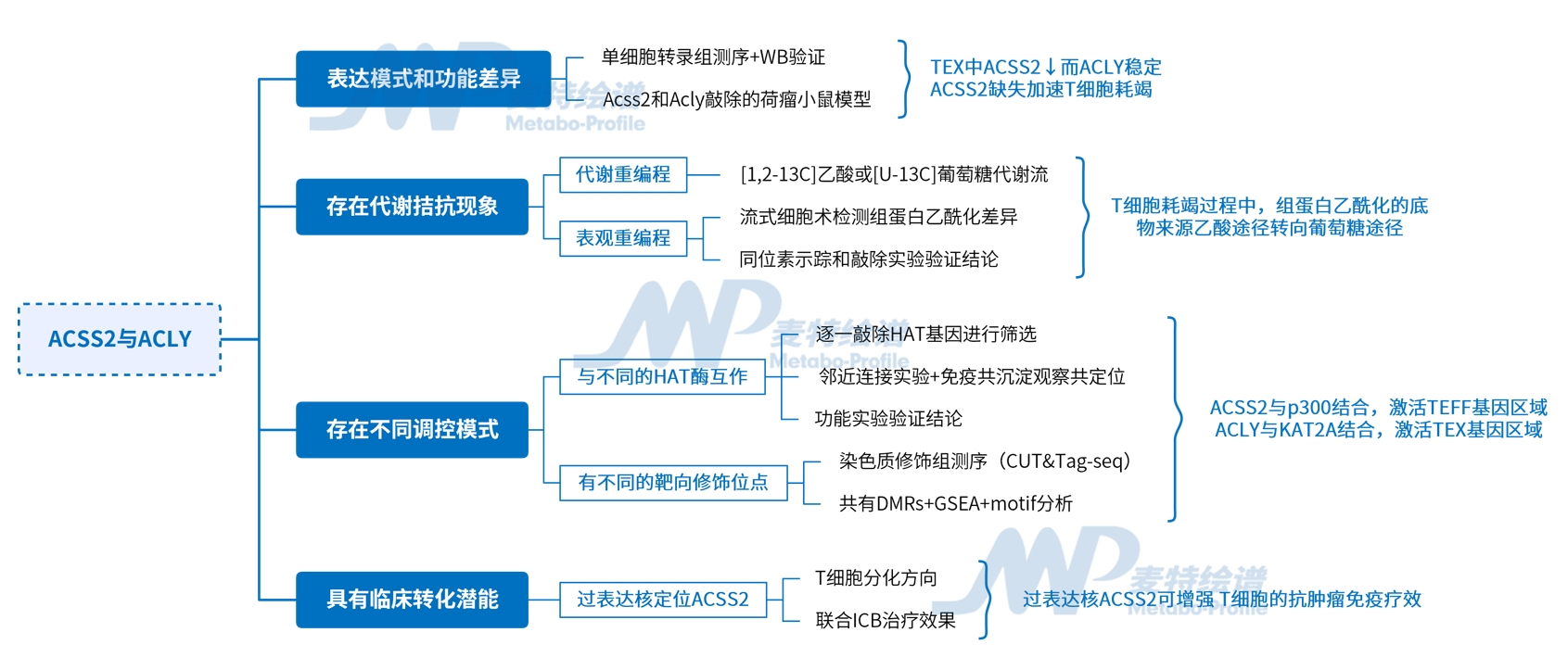
技術路線
研究結果
1. ACSS2與ACLY的表達模式和潛在功能均存在差異
首先,研究者通過單細胞轉錄組測序分析發現,功能完整的效應CD8+T細胞(TEFF)高表達代謝酶ACSS2,而功能耗竭的T細胞(TEX)中ACSS2顯著降低,其中TEXterm較TEXprog的ACSS2下調更為明顯,且Western blot檢測進一步驗證了這一差異。值得注意的是,另一代謝酶ACLY在各T細胞亞群中表達穩定,提示其與ACSS2的調控機制存在本質區別。
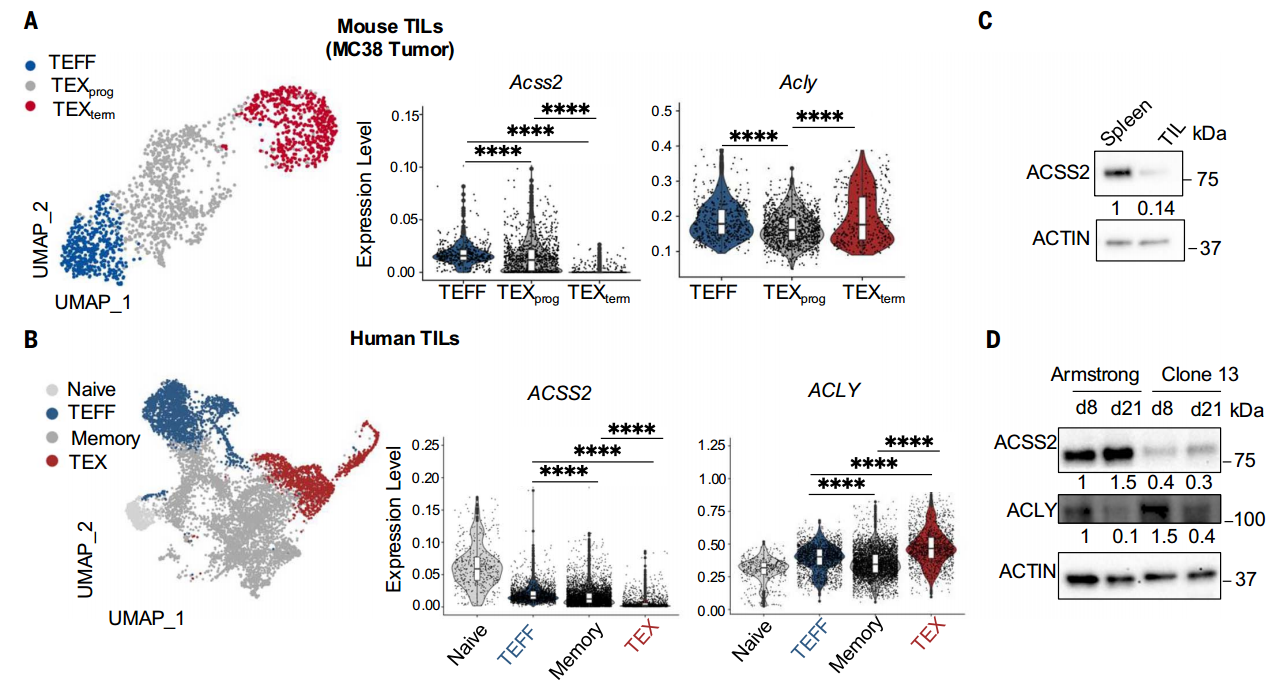
在TEX細胞中,ACSS2表達量減少,而ACLY維持不變
為明確二者的功能差異,研究者分別敲除TCR P14+CD8+T細胞的Acss2和Acly,然后過繼轉移至荷瘤及慢性感染小鼠模型,結果發現Acly缺陷型TILs表現出更強的腫瘤抑制能力,而Acss2缺失則導致T細胞完全喪失抗腫瘤活性。隨后的機制研究表明,Acly敲除促使TCF-1+TEXprog亞群顯著擴增,并伴隨IFN-γ+TNF+雙陽性效應細胞比例升高;而Acss2缺失組則加速向TEXterm分化,表現為效應分子表達下調及細胞因子分泌功能受損。
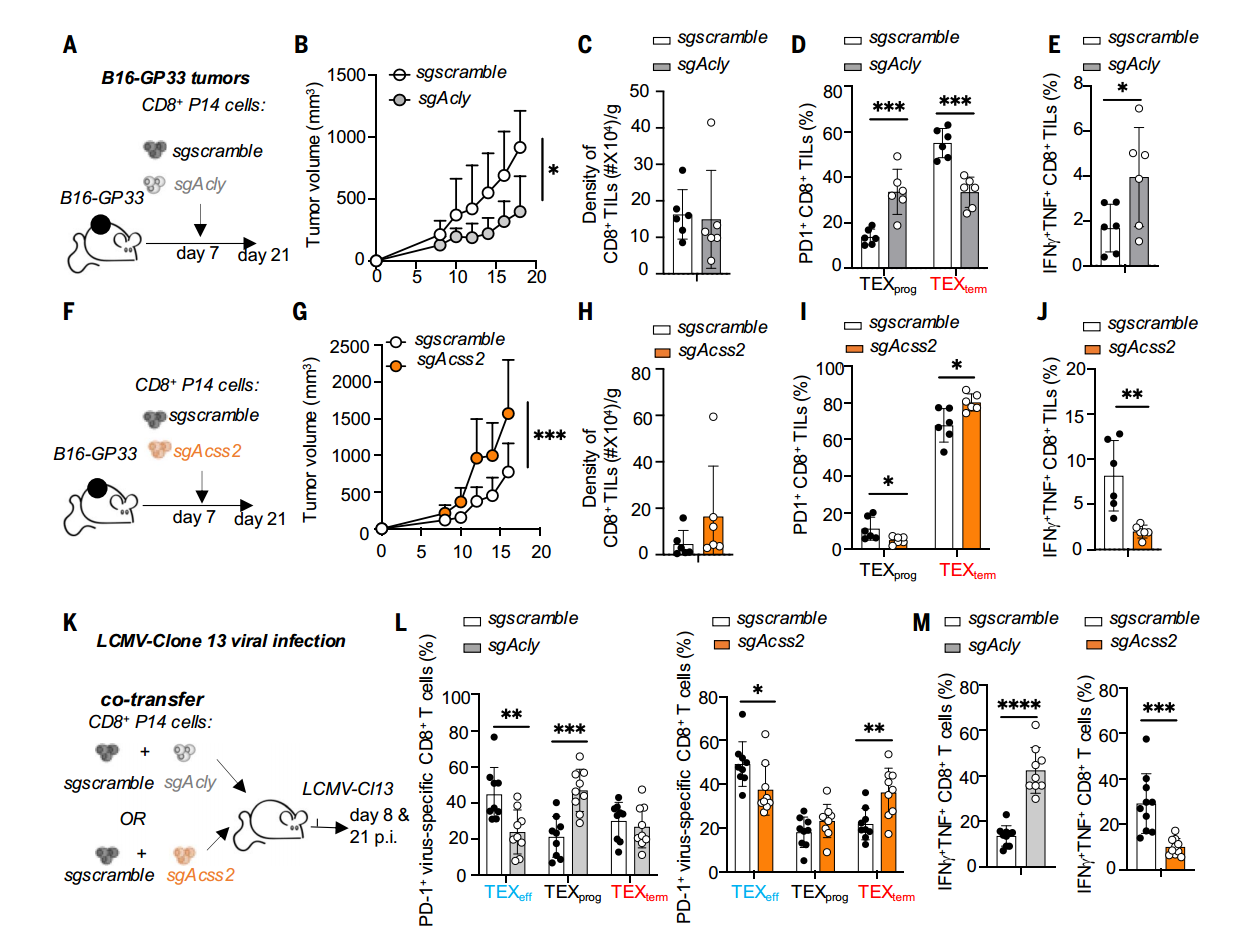
ACSS2促進TEXprog分化、抗腫瘤和抗病毒反應,而ACLY抑制
2. ACSS2與ACLY的代謝拮抗:乙酰輔酶A來源切換驅動表觀遺傳失衡
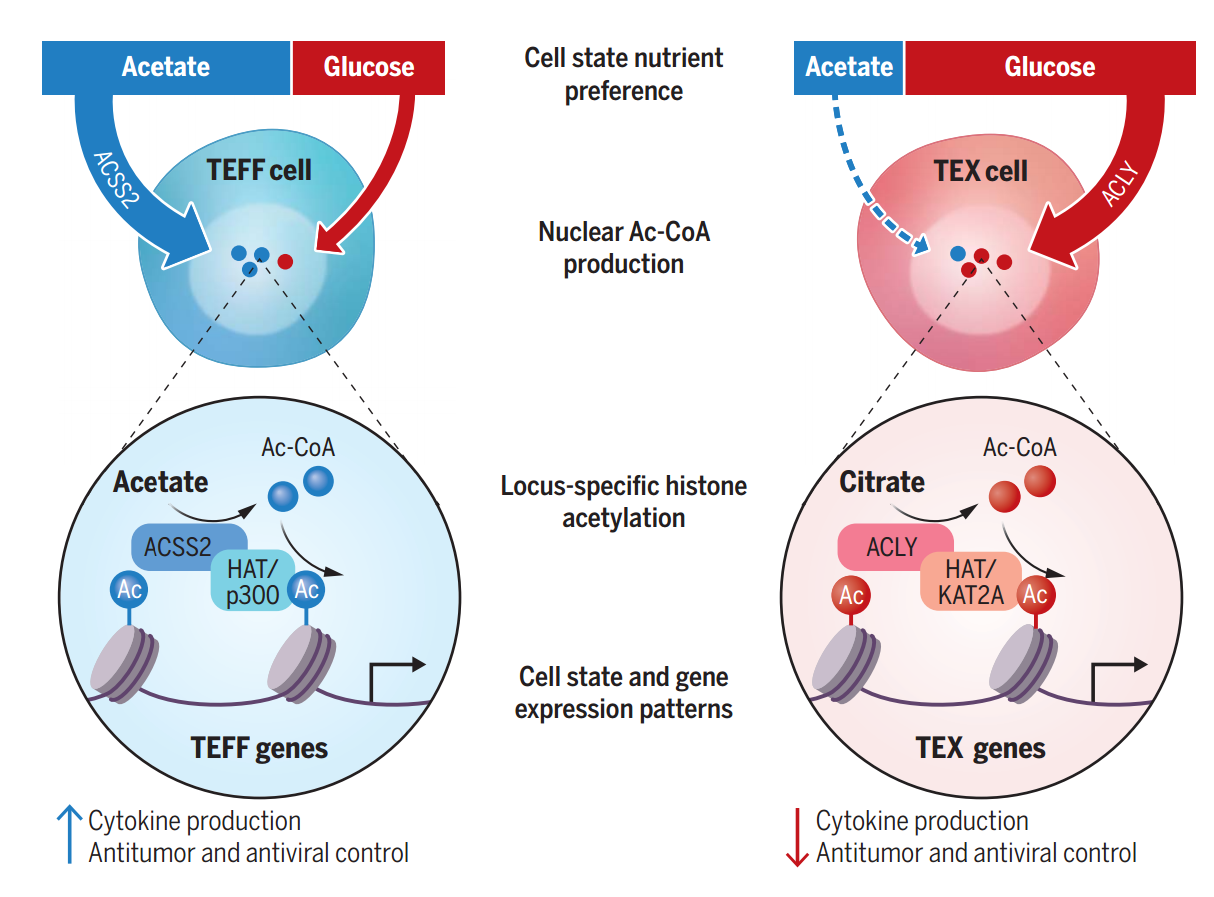
基于上述發現,研究者提出科學假設:功能正常的效應T細胞(TEFF)與耗竭T細胞(TEX)可能通過不同代謝途徑(乙酸或葡萄糖)生成乙酰輔酶A。為此進行了穩定同位素示蹤實驗,分別從急/慢性感染小鼠中分離TEFF與TEX細胞,使用[1,2-13C]乙酸或[U-13C]葡萄糖進行4小時穩態標記,通過質譜檢測代謝通量差異。結果顯示,相較于TEFF細胞,TEX細胞內乙酸來源的M2標記乙酰輔酶A顯著減少,而葡萄糖來源的標記比例升高,表明TEX細胞更依賴葡萄糖而非乙酸途徑生成核內乙酰輔酶A,這與ACSS2表達下調的特征一致。
由于乙酰輔酶A是蛋白質乙酰化的直接底物,研究者進一步解析了TEX與TEFF細胞的組蛋白乙酰化差異。流式細胞術檢測發現, TEX細胞的H3K27ac和H3K9ac水平較TEFF細胞顯著降低,且TEXterm的修飾水平也低于TEXprog。同位素示蹤實驗進一步揭示,TEFF細胞優先利用乙酸來源的乙酰輔酶A進行組蛋白乙酰化,而TEX細胞轉向依賴葡萄糖代謝供給。機制驗證實驗表明,敲除Acss2基因導致TEXprog細胞的H3K27ac水平下降50%,而Acly敲除對全局乙酰化無顯著影響,甚至因ACSS2代償性上調引發TEXterm細胞的H3K27ac水平回升。

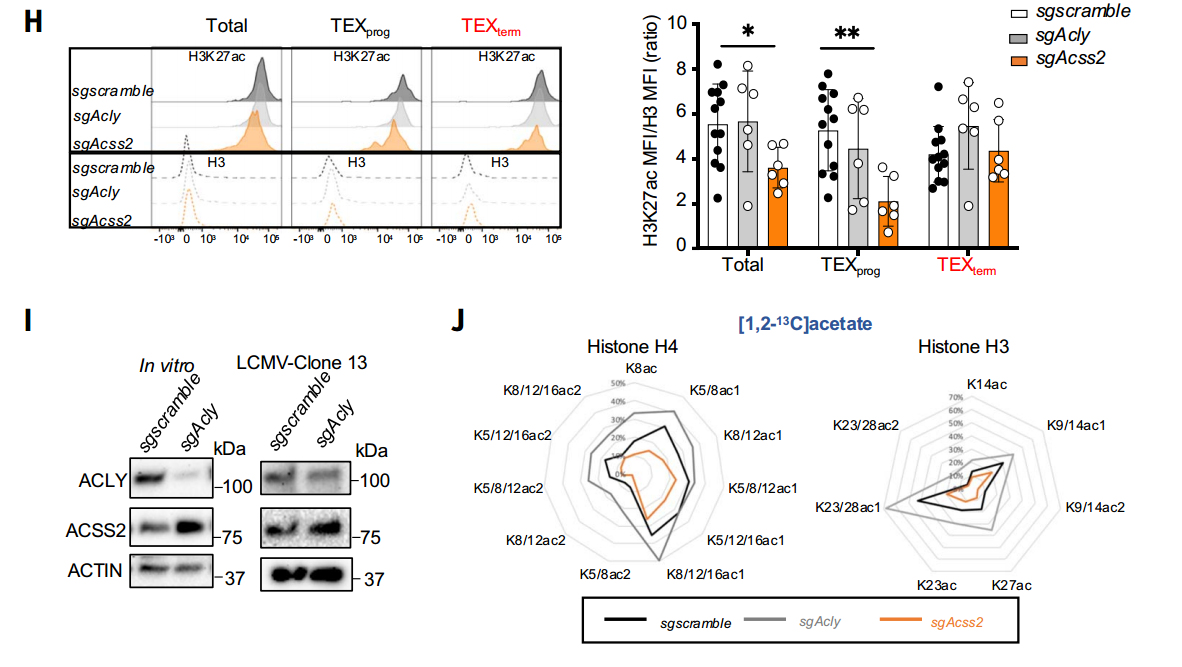
ACSS2和ACLY差異調節TEFF和TEX細胞之間的乙酰輔酶A產生和組蛋白乙酰化
這些數據共同表明,CD8+T細胞從TEFF向TEX(尤其是TEXterm)分化過程中,組蛋白乙酰化的代謝來源從ACSS2介導的乙酸途徑轉向ACLY依賴的葡萄糖途徑,但整體乙酰化水平仍呈進行性下降,提示代謝來源轉換無法完全補償表觀遺傳失衡。
3. ACSS2與ACLY的調控機制:通過HAT特異性互作靶向不同的乙酰化位點
通過系統性敲除CD8+T細胞中的HAT基因,研究者發現:敲除p300或CBP促進TEXterm分化,而敲除KAT2A則增強TEXprog形成。鄰近連接實驗(PLA)結合免疫共沉淀進一步證實,ACLY與KAT2A在TEX細胞中形成特異性互作,而ACSS2與p300在TEFF細胞中共定位。功能驗證實驗表明,ACSS2依賴p300維持TEXprog相關位點的組蛋白乙酰化(如H3K27ac),而ACLY通過KAT2A驅動TEXterm特征基因的乙酰化修飾。這種代謝酶-HAT復合物的空間特異性互作揭示了T細胞分化命運的分子開關。
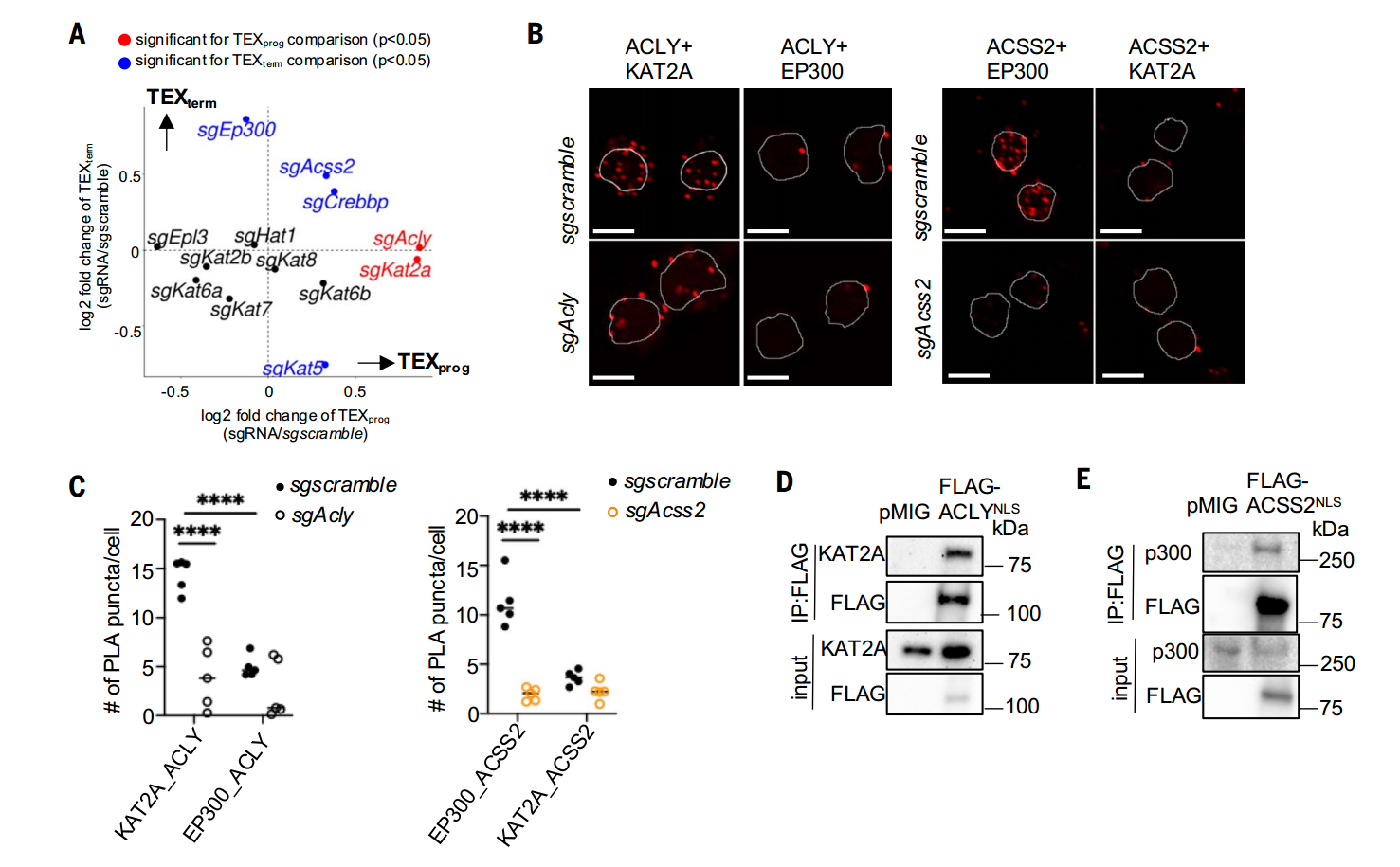
ACSS2和ACLY分別與p300和KAT2A相互作用
染色質修飾組分析(CUT&Tag-seq)表明代謝酶(ACSS2/ACLY)與HAT(p300/KAT2A)的復合物靶向不同染色質區域,從而調控T細胞向不同方向分化。
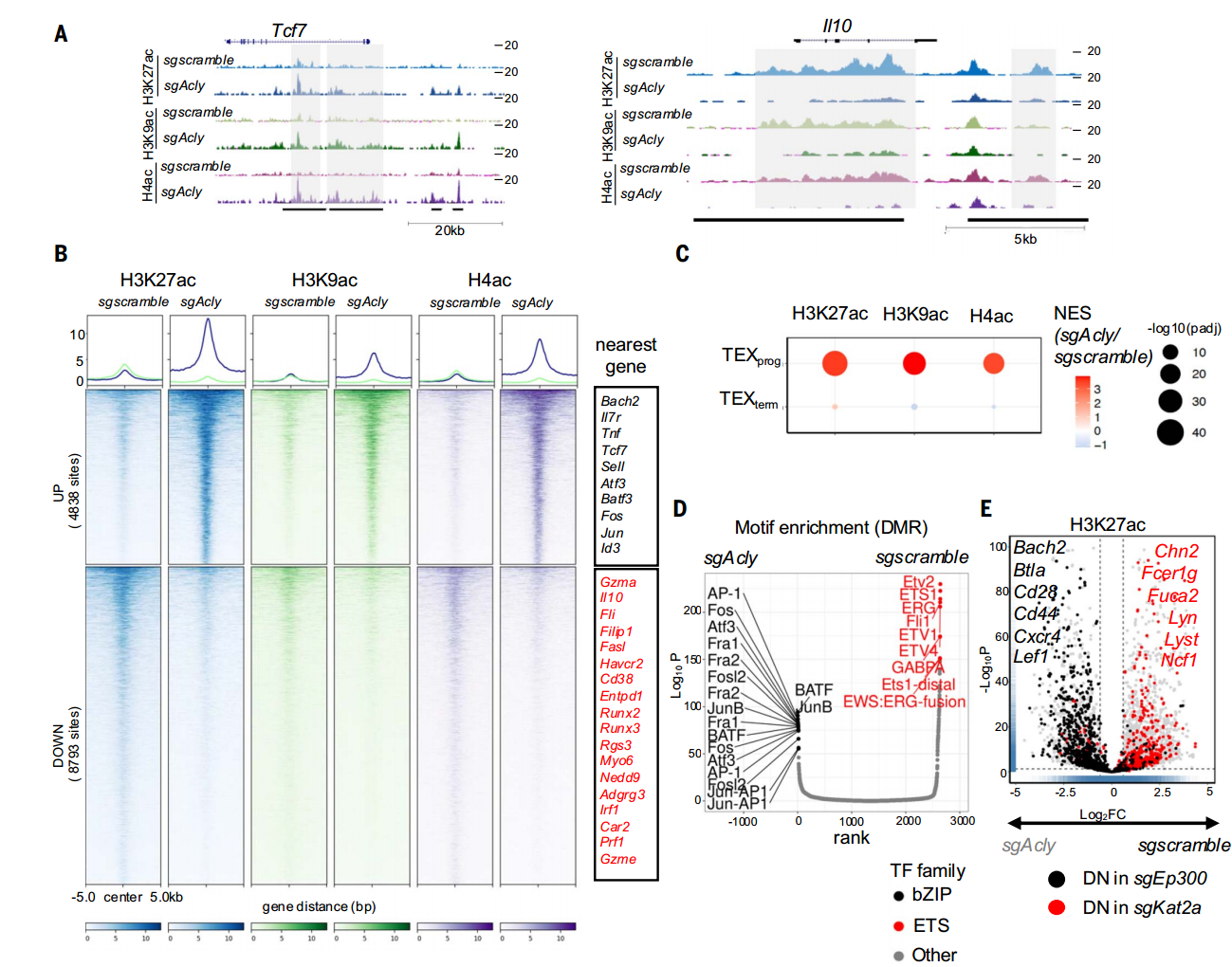
ACSS2和ACLY分別與p300和KAT2A協同調節基因座特異性組蛋白乙酰化
4. 臨床轉化潛能:核定位ACSS2增強T細胞干性,提升ICB療效
最后,研究者通過荷瘤小鼠模型發現,核定位ACSS2過表達T細胞(ACSS2NLS-OE)可顯著提升腫瘤浸潤淋巴細胞(TILs)的組蛋白乙酰化水平,特異性富集TEXprog基因(Tcf7/Bach2)位點的乙酰化修飾。轉錄組顯示ACSS2NLS-OE TILs中TEXprog基因上調而TEXterm基因受到抑制,證實核ACSS2通過表觀重塑維持T細胞干性。此外,臨床轉化研究發現,ACSS2NLS的過表達聯合ICB治療可增強細胞因子分泌,控制腫瘤生長;值得注意的是,抑制葡萄糖代謝(ACLY抑制劑BMS-303141)也可代償性上調ACSS2,協同ICB以CD8+T細胞依賴性方式顯著抑制腫瘤進展。
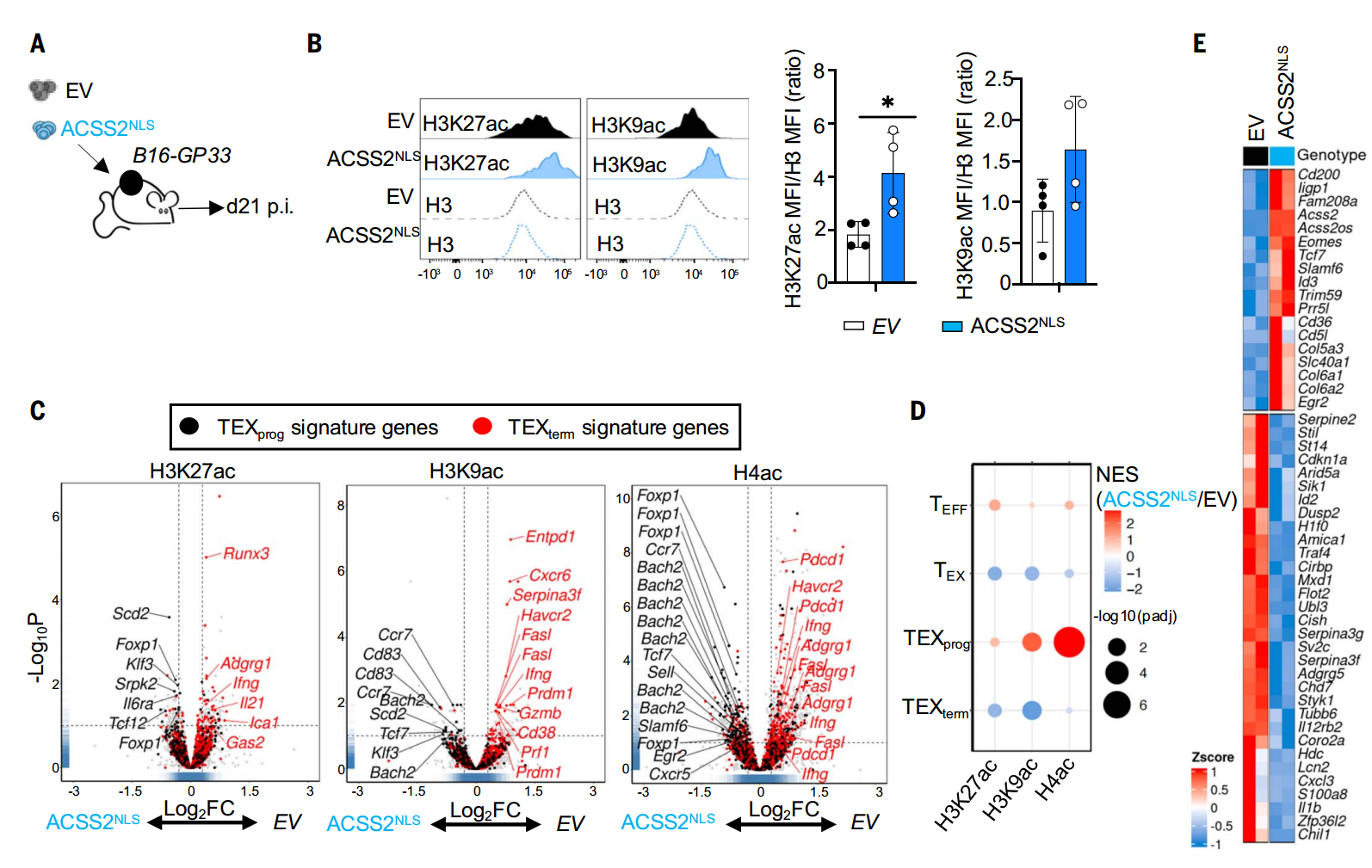
過表達ACSS2促進TEXCL2相關位點的組蛋白乙酰化并增強基因表達

過表達核ACSS2或抑制ACLY均可增強CD8+T細胞的抗腫瘤免疫
全文總結
本研究發現,代謝酶ACSS2與ACLY通過調控細胞核內乙酰輔酶A的生成,與組蛋白乙酰轉移酶(HATs)協同作用,以染色質位點特異性的方式決定CD8+T細胞的分化方向。核定位的ACSS2突變體能顯著增強干細胞樣耗竭前體細胞(TEXprog)的生成;而抑制ACLY活性,可將終末耗竭的TEXterm細胞重編程為TEXprog狀態。該研究不僅揭示了代謝與表觀遺傳互作在T細胞命運抉擇中的核心地位,還為聯合代謝干預(如ACLY抑制劑)與免疫檢查點阻斷提供了新思路,為優化過繼性T細胞治療策略奠定理論基礎。
參考文獻
Ma S, Dahabieh MS, Mann TH, et al. Nutrient-driven histone code determines exhausted CD8+T cell fates. Science. 2025
請掃描二維碼閱讀原文

繪譜幫您測
麥特繪譜提供全面的代謝流檢測服務,可追蹤含13C和15N等被標記物100+種,全面覆蓋糖酵解和TCA循環通路、磷酸戊糖途徑、 氨基酸代謝、脂肪酸代謝、 一碳代謝、 核苷酸代謝通路等。豐富的個性化標記定制經驗–[U-13C6]-Fructose,[U-13C16]-Palmitate, [U-13C3]-Serine,[U-13C2]-Glycine, [U-13C3]-Alanine, [U-13C3]-Pyruvate, [U-13C4]Malic Acid, [U-13C18]-Oleic Acid, 13CO2, 15N-NH4CL, [1, 2-13C2]-Glucose, [2,3,3-D3]-Serine, [2,3-13C2]Alanine, [1,2,3-13C3]-Choline等。歷經數年項目積累,檢測各類貼壁細胞、懸浮細胞、菌體、培養液、線粒體、組織、糞便等樣本類型,涵蓋多發性骨髓瘤、肝癌、線粒體遺傳代謝病、免疫細胞活性與疾病、心血管疾病等多個研究方向,合作項目成果突出,文章平均IF >10+。