文章標題:E3 ubiquitin ligase TRIM21-mediated K48-linked ubiquitination of ALDH2 rs671 mutant promotes adverse cardiac remodeling
合作客戶:山東大學齊魯醫院陳玉國、徐峰教授團隊
維真助力
|
基因信息 |
Aldh2:乙醛脫氫酶2 Trim21:一種E3連接酶 |
|
實驗動物 |
野生型和rs671小鼠 |
|
病毒產品 |
AAV9-Aldh2(1x10^13vg/ml); AAV9-CD68-mir30-shTrim21(6.75x10^13vg/ml) |
|
注射量 |
5x10^11vg |
|
注射方式 |
尾靜脈注射 |

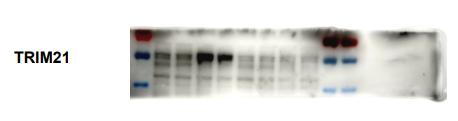
AAV病毒載體有效介導Aldh2的過表達及Trim21的沉默
研究背景
心梗后心衰是急性心肌梗死患者恢復期的首要死亡原因,盡管再灌注治療可恢復心肌血供,但心梗后心衰的死亡率和致殘率仍居高不下。ALDH2 rs671是人類常見的單核苷酸突變,與心梗后主要不良心臟事件(MACE)風險升高相關,然而該突變影響心梗后心臟重構的分子機制尚不明確。蛋白泛素化是心衰發病的關鍵調控機制,但ALDH2 rs671突變體的泛素化修飾尚未被闡明,亟需鑒定其上游E3泛素連接酶并明確作用機制。
部分研究結果
1、TRIM21介導ALDH2 rs671泛素化降解致心梗后心臟不良重構
研究團隊通過構建骨髓嵌合小鼠心梗模型,發現攜帶ALDH2 rs671突變的小鼠在MI后出現更嚴重的心肌纖維化,心臟收縮功能顯著下降以及巨噬細胞胞葬作用缺陷;且突變組與野生組的心肌駐留/浸潤巨噬細胞數量無顯著差異,說明表型由巨噬細胞功能異常而非數量變化導致。組織和細胞實驗證實,ALDH2 rs671 突變體的蛋白水平顯著降低,但mRNA水平與野生型無差異;在心肌梗死患者和小鼠的缺血組織中,ALDH2蛋白水平進一步下調。環己酰亞胺蛋白合成抑制實驗顯示,ALDH2 rs671突變體的降解速率顯著快于野生型;使用蛋白酶體抑制劑MG132處理可顯著抑制其降解,而氯喹無作用,證實其經蛋白酶體途徑降解。進一步的研究發現TRIM21是ALDH2的E3泛素連接酶,TRIM21通過K48位點連接的泛素化修飾ALDH2,主要作用于K73位點;ALDH2 rs671突變體與TRIM21結合更強,導致其更易被降解。
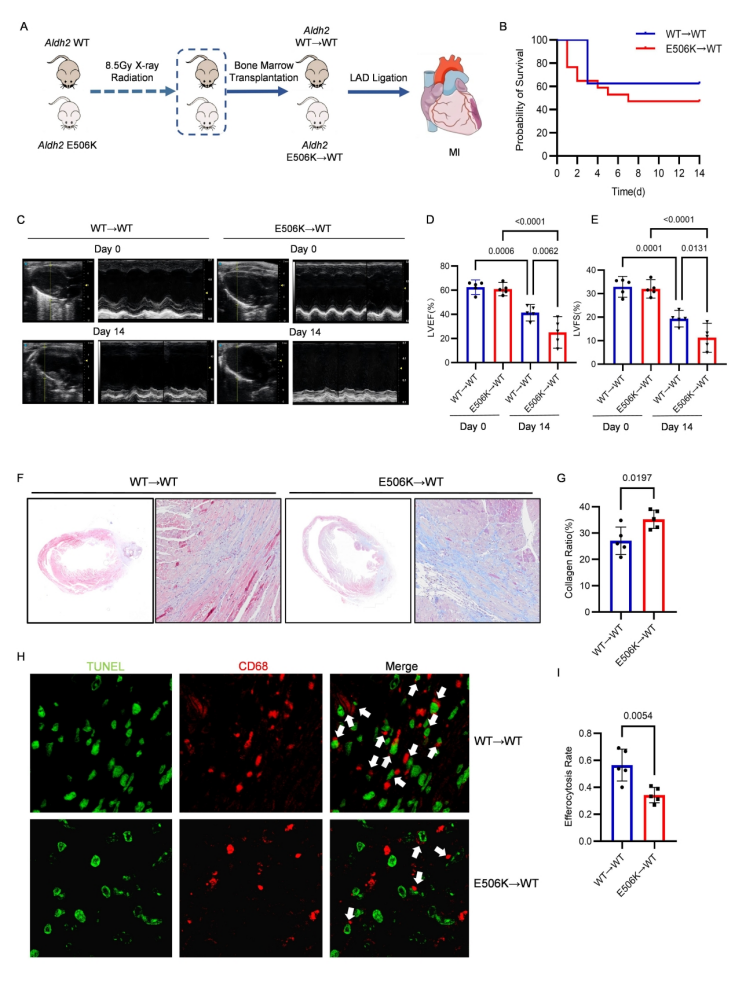
骨髓特異Aldh2 rs671變異增強心肌梗死后心臟纖維化
2、靶向干預TRIM21/ALDH2可逆轉心梗后心臟不良重構
為進一步驗證ALDH2的功能,研究團隊使用AAV9-Aldh2進行拯救實驗,發現ALDH2的過表達能夠減輕MI后心功能的下降幅度以及心臟修復過程中的心臟纖維化,同時恢復巨噬細胞胞葬功能。為闡明TRIM21在MI后心臟纖維化過程中巨噬細胞中的作用,使用CD68啟動子驅動的AAV構建了巨噬細胞特異性Trim21敲低小鼠,結果顯示敲低Trim21恢復了ALDH2 rs671突變導致的巨噬細胞胞葬功能缺陷,顯著提高心梗后小鼠生存率,逆轉心肌纖維化,恢復心臟收縮功能。這些結果表明,通過下調Trim21,rs671突變介導的心臟纖維化缺陷得到改善。
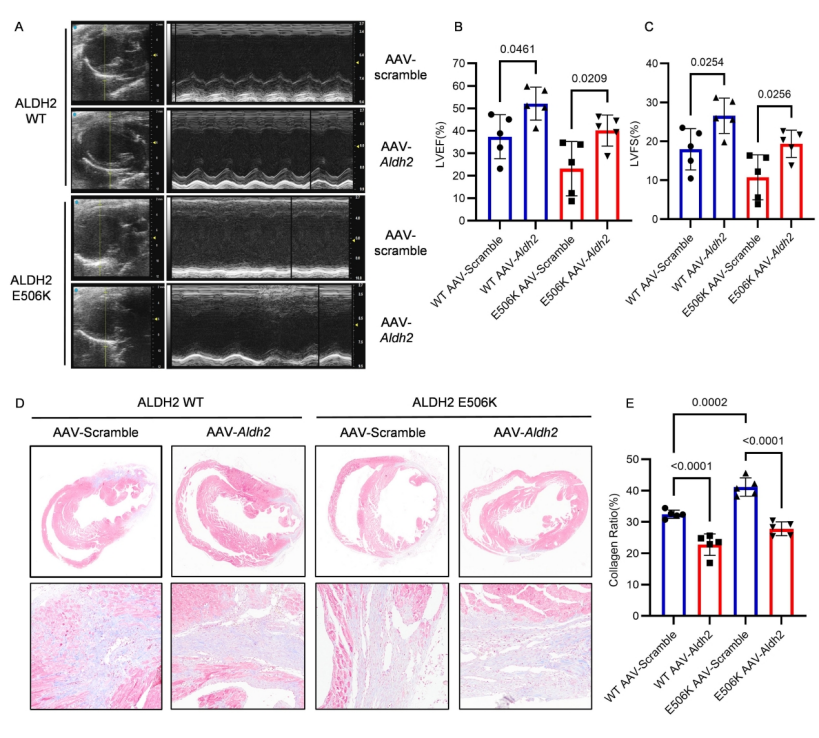
Aldh2過表達挽救了由Aldh2 rs671變體誘導的心臟功能惡化
研究結論
本研究首次發現E3泛素連接酶TRIM21是ALDH2 rs671 突變體的上游調控因子,其可催化ALDH2第73位賴氨酸發生K48位點連接的泛素化并加速該突變體的蛋白酶體降解,導致巨噬細胞的胞葬功能缺陷,進而加劇急性心肌梗死后的心肌纖維化和心臟收縮功能障礙。證實TRIM21是心肌梗死后心力衰竭的潛在治療靶點,且揭示了TRIM21-ALDH2-Rac2-胞葬作用調控軸在心梗后心肌纖維化中的關鍵調控機制。