血管損傷誘導的再狹窄是冠心病患者長期預后不良的重要原因,盡管乙醛脫氫酶2(ALDH2)缺乏與冠心病患者預后不良有關,但ALDH2影響血管損傷誘導的再狹窄的確切機制仍不清楚。2025年10月,山東大學齊魯醫院陳玉國/徐峰教授團隊在Metabolism(IF11.9)發文“ALDH2 deficiency aggravates vascular injury-induced restenosis by enhancing vascular smooth muscle cell proliferation through SLC38A2-mediated upregulation of glutamine uptake”,研究結果闡明了ALDH2 缺陷會通過增加谷氨酰胺攝取來促進血管平滑肌細胞增殖,進而加劇新生內膜形成,這一發現為預防血管損傷誘導的再狹窄提供了一種具有潛力的轉化策略。

· 維真助力 ·
In vivo
基因信息
ALDH2:乙醛脫氫酶2
SLC38A2:溶質載體家族38成員2
實驗動物
8周齡雄性ALDH2flox和ALDH2ΔSMC小鼠;C57BL/6小鼠
病毒產品
AAV2-SM22α-GFP-mir30-sh-SLC38A2、 AAV2-sh-Scramble;
AAV2-SM22α-GFP-ALDH2、AAV2-GFP
病毒用量
5×1011
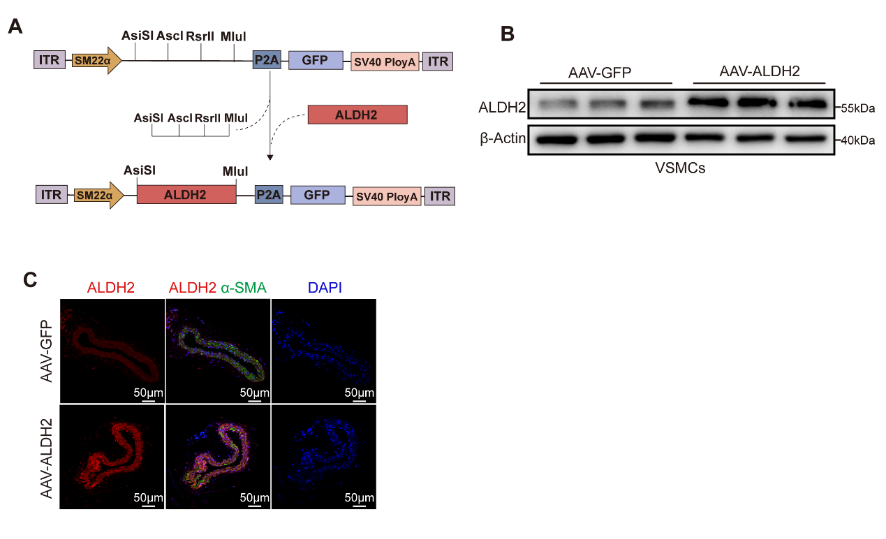
SMC特異性ALDH2過表達AAV的構建與轉染效率驗證
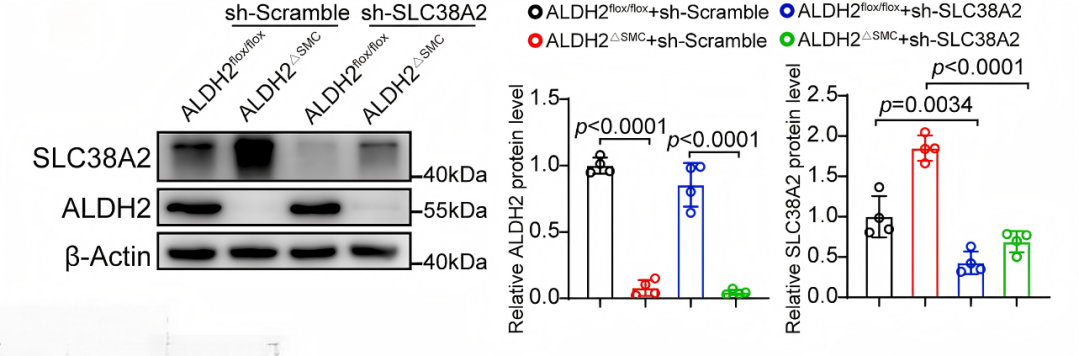
蛋白質印跡證實MASMC中SLC38A2表達被顯著抑制
In vitro
病毒產品
Lv-shALDH2、Lv-Control
感染細胞
原代小鼠主動脈平滑肌細胞MASMCs
研究結果
1.ALDH2缺陷促進新生內膜形成
研究團隊發現在人狹窄冠狀動脈組織的新生內膜區域、小鼠股動脈導絲損傷模型及頸動脈結扎模型的損傷血管VSMCs中,ALDH2顯著下調。在體內實驗中,全身性ALDH2敲除與VSMC特異性ALDH2敲除均會促進血管損傷后的新生內膜形成。研究人員使用攜帶SM22α啟動子的AAV2在小鼠血管平滑肌細胞中特異性過表達ALDH2,以AAV-GFP組作為對照。通過Western blot和免疫熒光染色證實,AAV-ALDH2組小鼠血管中的ALDH2蛋白表達水平顯著高于對照組。與對照組相比,ALDH2過表達顯著減輕兩種模型中的新生內膜形成,并有效抑制損傷動脈中的VSMC增殖。隨后研究人員從ALDH2基因敲除小鼠和野生型小鼠中分離原代小鼠主動脈平滑肌細胞,探索ALDH2在PDGF-BB中的潛在作用,結果表明ALDH2敲除細胞遷移及增殖能力增強,使用siRNA敲低MASMCs中的ALDH2,同樣可以增強PDGF-BB誘導的VSMC增殖和遷移。作為反向驗證,研究人員在MASMCs中通過慢病毒載體過表達ALDH2,證實VSMC遷移和增殖受到抑制。
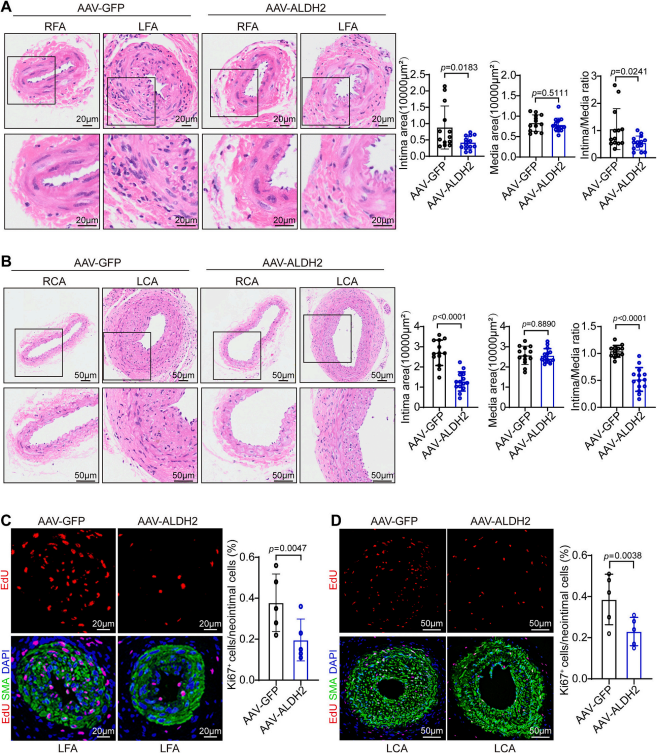
ALDH2過表達通過抑制VSMCs增殖減少新生內膜形成
2.ALDH2缺陷通過上調SLC38A2的表達促進VSMC增殖
mRNA測序和代謝組學分析顯示,在ALDH2敲除的VSMCs中,谷氨酰胺和谷胱甘肽的水平顯著升高,并且谷胱甘肽代謝通路被富集。對谷氨酰胺轉運蛋白進行篩選后發現,SLC38A2在mRNA和蛋白水平上均被ALDH2缺陷特異性上調,而其他轉運蛋白未受影響。在VSMCs中過表達SLC38A2,能夠模擬ALDH2敲除的表型,即促進PDGF-BB誘導的細胞增殖、遷移并激活mTORC1信號通路。在ALDH2敲低的VSMCs中,使用siRNA敲低SLC38A2,可以有效逆轉由ALDH2缺陷引起的mTORC1信號激活、細胞增殖和遷移增強。研究團隊探索了ALDH2上調SLC38A2表達的分子機制,通過mRNA-seq數據與Jaspar數據庫預測的轉錄因子進行交叉比對,篩選出13個潛在調控SLC38A2的轉錄因子。其中,激活轉錄因子4(ATF4)表達量最高,且實驗證實ALDH2敲低會顯著上調ATF4的表達水平。進一步的研究發現ALDH2缺陷激活轉錄因子ATF4,使其與SLC38A2基因啟動子的結合增加,從而驅動SLC38A2的轉錄,體外數據表明ATF4是ALDH2-SLC38A2介導的谷氨酰胺攝取和VSMC增殖所必需的。
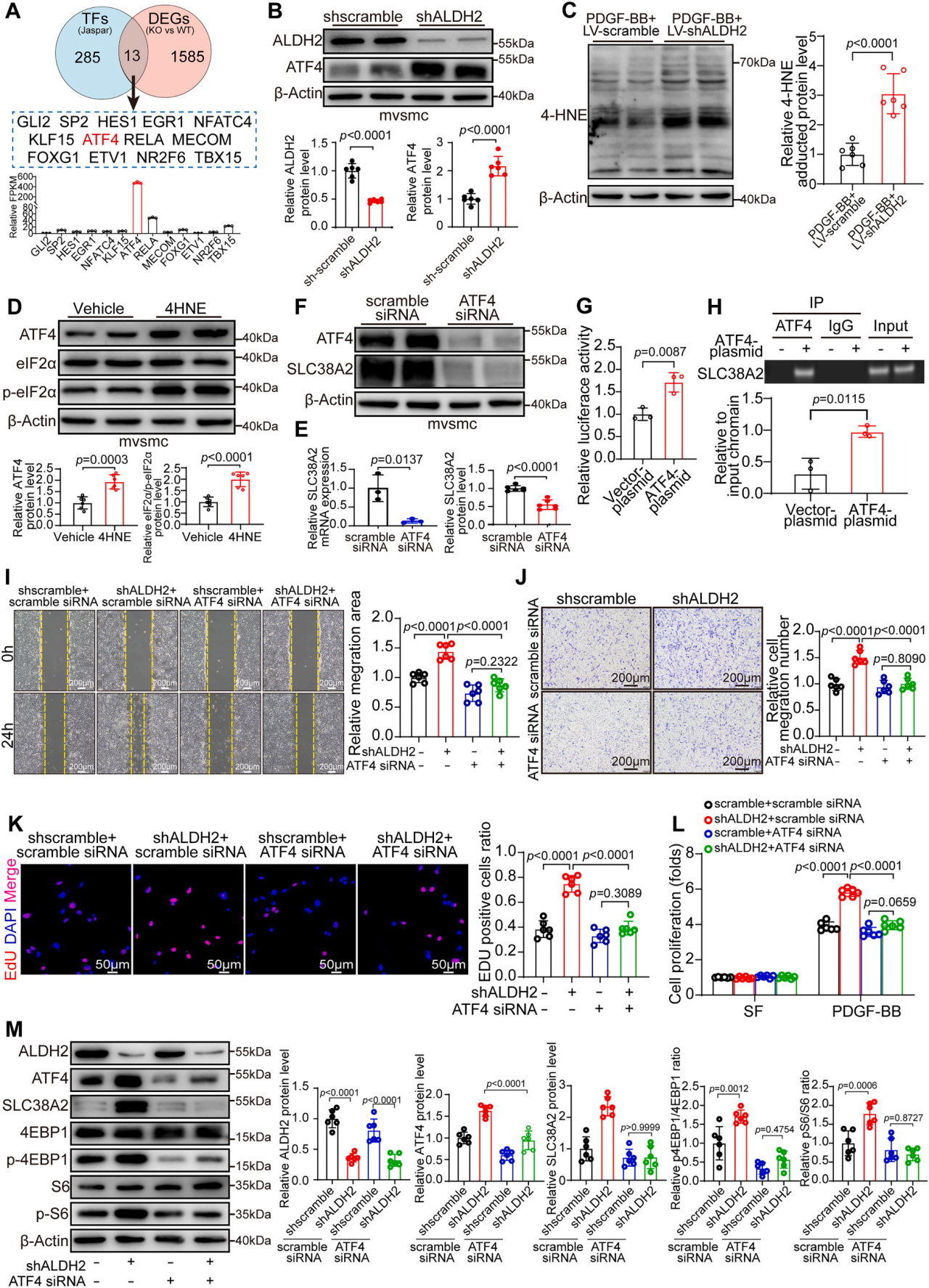
ALDH2缺失上調SLC38A2并通過ATF4介導的轉錄激活促進VSMCs增殖和遷移
3.SLC38A2是ALDH2調控新生內膜形成的下游關鍵執行者
使用AAV2-SM22α-shSLC38A2注射VSMC特異性ALDH2敲除小鼠及其對照小鼠中,實現VSMC特異性SLC38A2敲低,四周后對小鼠進行股動脈導絲損傷和頸動脈結扎手術,以誘導血管新生內膜形成。研究數據顯示,ALDH2敲除小鼠新生內膜面積和內膜/中膜面積比顯著增加,敲低SLC38A2明顯減輕上述新生內膜形成。Ki67免疫熒光染色顯示,在VSMC特異性ALDH2缺失組中,損傷動脈內增殖的Ki67陽性細胞數量的增加,可被SLC38A2缺失所消除。這些結果證明,ALDH2缺陷通過SLC38A2促進血管平滑肌細胞增殖及損傷誘導的新生內膜形成。進一步探究發現ALDH2的酶活性是其發揮保護作用的核心,其功能喪失會直接加劇疾病。

VSMC特異性SLC38A2敲低減少了ALDH2缺失引起的新生內膜形成
研究結論
本研究闡明了一種新機制:ALDH2通過激活溶質載體家族38成員2(SLC38A2)-谷氨酰胺攝取軸,調控血管平滑肌細胞增殖與新生內膜形成。ALDH2-ATF4-SLC38A信號通路的發現,不僅加深了對ALDH2介導細胞行為的分子機制的理解,也為治療血管損傷誘導的再狹窄提供了潛在的靶點。