心肌肥厚是導致心力衰竭和心臟性猝死的主要原因之一,OTUD7a是一種去泛素化酶,也是一種潛在的腫瘤抑制因子,此前被證實與神經發育障礙和腫瘤相關,其對心血管疾病尤其是心臟肥大的作用尚未闡明。2025年11月21日,鄭州大學第一附屬醫院王小芳/鄭哲團隊在Circulation Research (IF 16.2)在線發表文章“OTUD7a Accelerates Pathological Cardiac Hypertrophy via TAK1 Activation”,研究首次證實OTUD7a 是病理性心肌肥厚的新型促進因子,并表明靶向OTUD7a-TAK1軸是治療心肌肥厚及相關心力衰竭的潛在有效策略。

· 維真助力·
基因信息 OTUD7a:OTU結構域包含蛋白7a
感染細胞 新生大鼠心肌細胞
病毒產品 Ad-OTUD7a
MOI 50
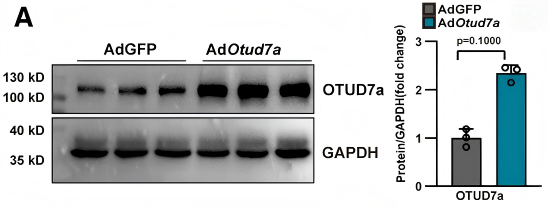
Ad-OTUD7a轉導的新生大鼠心肌細胞中OTUD7a成功過表達
研究結果
1、體內外實驗證實OTUD7a促進病理性心肌肥厚,調控心臟重塑與心功能
研究結果證實OTUD7a在體內TAC手術和體外苯腎上腺素誘導的心肌肥厚模型中表達上調,全身性敲除OTUD7a可減輕小鼠壓力超負荷誘導的心臟功能障礙與結構重塑,RNA測序及基因集富集分析顯示OTUD7a調控的基因在纖維化、心臟功能和蛋白質加工通路中富集,提示OTUD7a可能參與這些生物學過程來影響心臟重塑。隨后構建OTUD7a雜合敲除小鼠模型,證實雜合敲除OTUD7a減輕了TAC誘導的小鼠心肌重構與心功能障礙。進一步在心肌組織特異性敲除及過表達OTUD7a以確定其在體內心臟重塑和功能障礙中的功能作用,結果顯示心肌特異性缺失OTUD7a可減輕小鼠病理性心肌肥厚并改善心功能障礙,過表達OTUD7a則產生相反的結果。體外驗證實驗中以新生大鼠心肌細胞為研究對象,PE刺激成功構建體外心肌肥厚模型,通過腺病毒實現OTUD7a過表達或沉默,結果表明OTUD7a在體外可調控去氧腎上腺素(PE)誘導的心肌細胞肥厚,過表達OTUD7a促進該病理過程,沉默則抑制。
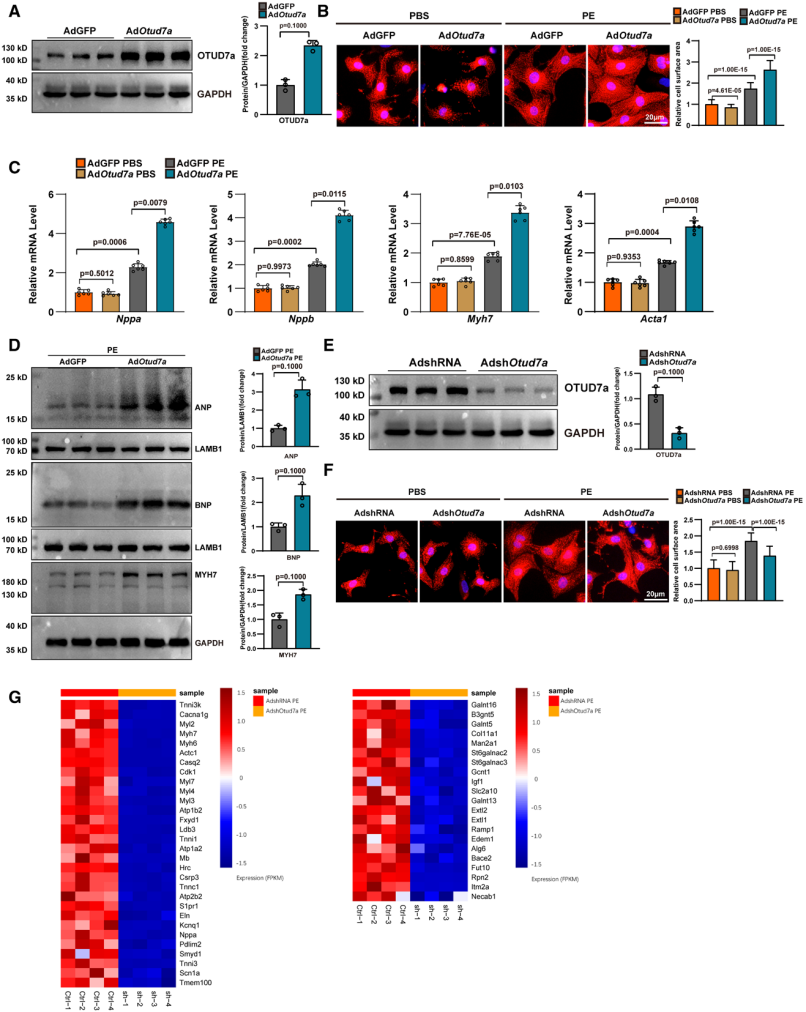
OTUD7a在體外調節去氧腎上腺素誘導的心臟肥大
2、OTUD7a通過靶向激活TAK1-JNK-P38信號通路,調控病理性心肌肥厚
RNA測序及KEGG分析均顯示OTUD7a敲除小鼠心臟組織、OTUD7a沉默的心肌細胞中,MAPK信號通路為差異富集最顯著的通路,提示OTUD7a與該通路密切相關。與WT-TAC小鼠相比,OTUD7a敲除-TAC小鼠中p-JNK和p-P38的水平下調;與AAV9-GFP-TAC小鼠相比,OTUD7a過表達小鼠中,p-JNK、p-P38水平顯著升高;體外驗證結果與其一致。體內外分子機制上的探索實驗均證實,OTUD7a敲除/沉默可降低TAK1的總蛋白水平及p-TAK1水平;OTUD7a過表達則顯著升高TAK1及 p-TAK1水平。這些數據闡明了OTUD7a誘導的惡化的病理性心臟肥大可能依賴于TAK1-JNK-P38通路的激活。TAK1-JNK-P38信號通路的關鍵分子(TAK1、p-TAK1、p-JNK、p-P38)表達呈時間依賴性激活,且該趨勢在體外PE誘導的心肌細胞肥厚模型和體內TAC誘導的小鼠心肌肥厚模型中均一致。進一步使用TAK1抑制劑5Z-7-oxozeaenol可阻斷OTUD7a過表達引起的TAK1-JNK/P38通路激活,同時逆轉OTUD7a過表達導致的心肌肥厚加重,證實OTUD7a對心肌肥厚的調控依賴TAK1-JNK-P38通路。

OTUD7a可以調節蛋白質加工和MAPK信號傳導
3、OTUD7a直接結合TAK1促進心臟肥大
免疫共沉淀實驗及GST Pull-down實驗證實OTUD7a和TAK1蛋白存在相互作用。接著,研究團隊構建了OTUD7a和TAK1的系列截短體,確定了OTUD7a上第201至384位氨基酸組成的結構域與TAK1上第1至390位氨基酸組成的結構域發生結合,且二者共定位在細胞質中。進一步驗證發現OTUD7a 突變體無法激活TAK1,也不能促進心肌肥厚,證實OTUD7a對TAK1的調控依賴其完整功能結構域,此外OTUD7a過表達以濃度依賴方式上調 293T 細胞內源性TAK1蛋白水平。在PE刺激的NRCM中加入不同通路抑制劑,發現蛋白酶體抑制劑MG132可逆轉OTUD7a沉默引起的TAK1表達降低,而溶酶體抑制劑無挽救作用。為深入闡明OTUD7a穩定TAK1蛋白的機制,研究人員檢測了TAK1的泛素化水平,發現OTUD7a表達降低時,TAK1的泛素化水平顯著升高;OTUD7a過表達時,TAK1的泛素化水平顯著降低。體外泛素化實驗結果顯示,OTUD7a可對TAK1進行去泛素化修飾。上述研究結果表明TAK1的降解由OTUD7a通過K48連接的泛素化途徑介導。

OTUD7a穩定TAK1蛋白
研究結論
綜上,本研究首次證實OTUD7a是病理性心肌肥厚的重要致病因子。在心臟應激狀態下,OTUD7a直接與TAK1相互作用,抑制TAK1的泛素化降解,進而升高TAK1及其下游JNK/P38的磷酸化水平。OTUD7a通過TAK1-JNK-P38軸調控心肌肥厚的發生發展,這些發現為闡明病理性心肌肥厚的發病機制提供了新視角,并為心肌肥厚的治療提供了新策略。