研究背景
結直腸癌(CRC)是全球第三大常見癌癥,盡管免疫療法的進步給癌癥治療帶來了革命性變化,但其在CRC中的有效性仍然有限。已有研究表明APC驅動的多克隆性顯著增強了致瘤潛力,但APC突變在介導免疫逃避中的作用仍知之甚少。近期南方醫科大學周偉杰團隊在Cell Research (IF 25.9)上發表文章“Targeting PTPN13 with 11-amino-acid peptides of C-terminal APC prevents immune evasion of colorectal cancer”,揭示了一種以前未知的APC/PTPN13/STAT1依賴的腫瘤免疫抑制機制。
 維真助力 - 腺病毒·
維真助力 - 腺病毒·
病毒產品 Ad-Cre、Ad-GFP
實驗細胞 Apcfl/fl /LSL-KrasG12D/p53fl/fl小鼠腸道類器官
結果展示
1. APC缺失損害IFNγ-STAT1-IRF1信號傳導并抑制抗原呈遞
作者發現APC缺失損害CD8+T細胞浸潤誘導CRC的免疫逃避,提高CRC模型對免疫檢查點封鎖的抵抗力,且PTPN13介導APC缺失驅動的CRC免疫逃避。作者通過RNA-seq研究了CRC中APC缺失誘導的免疫逃避所涉及的信號通路,發現APC敲低與II型干擾素和JAK-STAT信號通路反應的抑制有關,Apc敲低顯著降低了IFNγ誘導的STAT1磷酸化和IRF1上調,而JAK1磷酸化保持不受影響,表明APC在該通路中在JAK1下游和STAT1上游發揮作用。作者還探究了APC缺失對抗原呈遞和CD8+T細胞功能的影響,發現Apc敲低后抗原呈遞受損,抗原特異性CD8+T細胞顯著減少。機制研究發現Stat1R274Q和Irf1的過度表達顯著抑制WT Balb/c小鼠皮下移植的CT26-shApc細胞的生長,并增強CD8+T細胞浸潤,這些結果表明,APC丟失通過抑制STAT1/IRF1信號傳導促進免疫逃避,而恢復該通路可有效抵消腫瘤免疫抵抗。
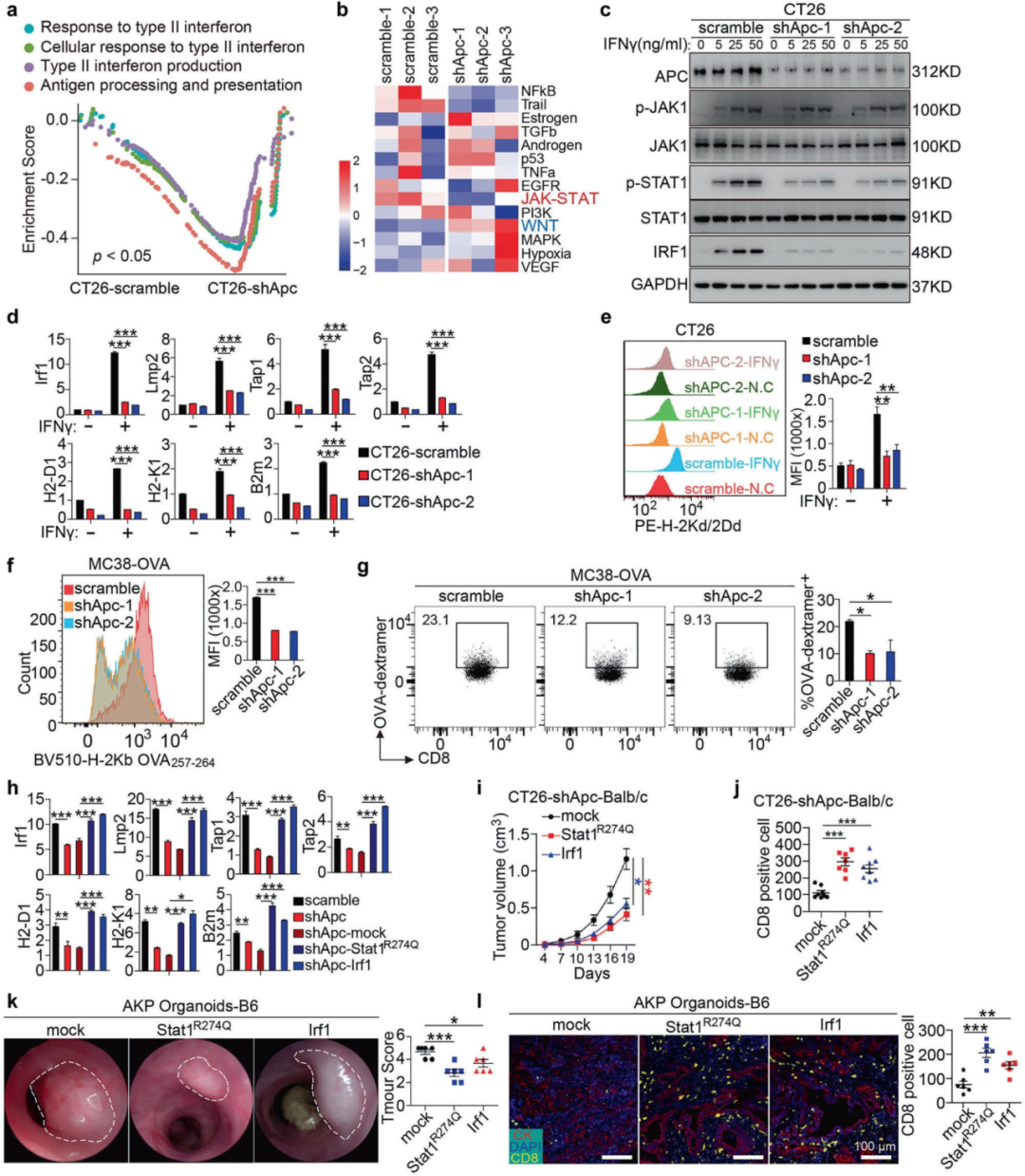
圖1 APC缺失使IFNγ-STAT1-IRF1-MHC-I抗原呈遞信號失活
2. PTPN13與STAT1的直接相互作用和去磷酸化
已有研究表明PTPN13參與了STAT家族成員的去磷酸化,作者通過免疫共沉淀確認STAT1–PTPN13存在相互作用,并證實STAT1直接結合到PTPN13的PDZ2a結構域。體外PTPase測定表明STAT1是PTPN13的直接底物,PTPN13使STAT1去磷酸化。免疫共沉淀還表明APC-PTPN13存在相互作用,并且IFNγ刺激增強了該復合物的形成,因此作者假設APC阻斷STAT1-PTPN13相互作用,這一觀點在Apc敲除細胞中增強的PTPN13-STAT1相互作用所證實。進一步研究發現APC的C端纈氨酸對于其與PTPN13的相互作用至關重要,并且這種相互作用對于調節CRC的免疫逃避至關重要。
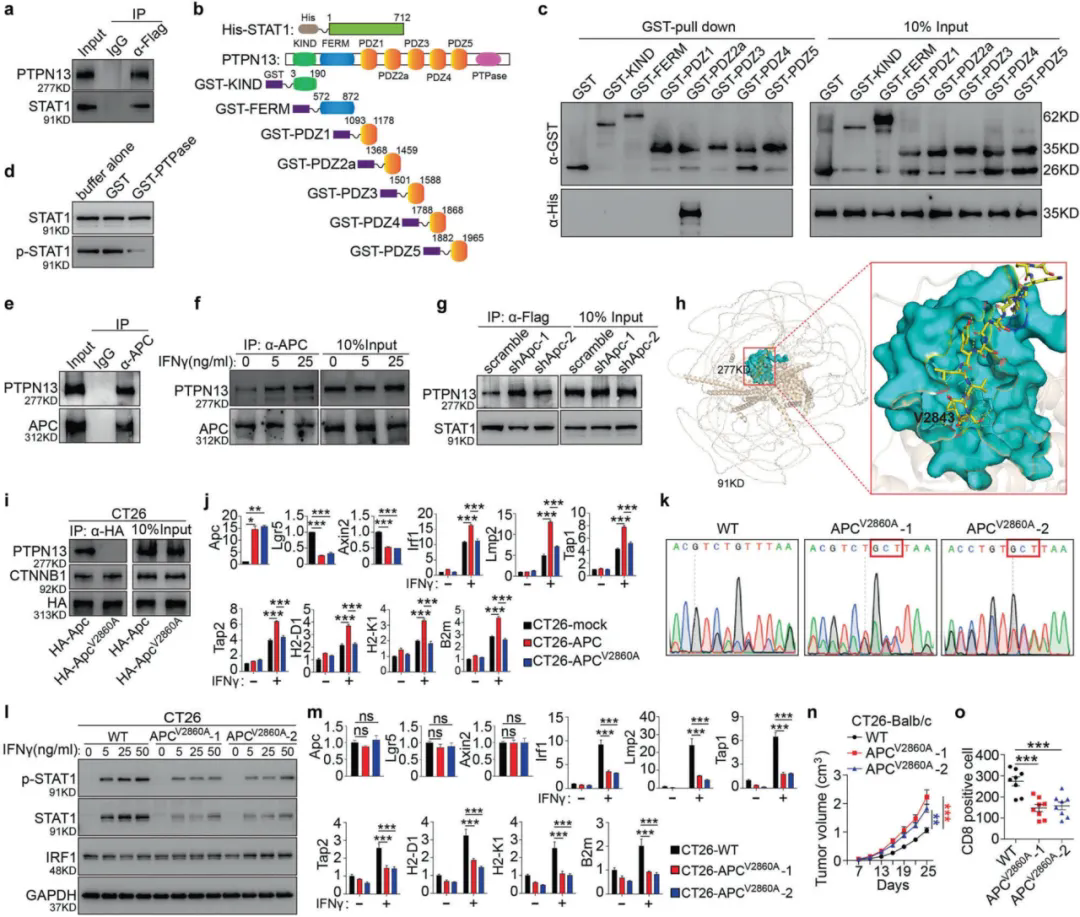
圖2 APC的C端纈氨酸對于CRC中PTPN13的結合和免疫逃避至關重要
3. APC11抑制腫瘤生長并增強抗PD1治療的反應
作者發現APC11(APC的C端最后11個氨基酸)有效阻斷PTPN13/STAT1相互作用,恢復APC缺失引起的IFNγ-STAT1-IRF1-MHC-I抗原呈遞途徑失活。TAT綴合APC11肽(TAT-APC11)治療顯著提高了腫瘤小鼠的生存率。為了提高溶解度和穩定性,作者將TAT-APC11與PEG綴合,評估了PEG-TAT-APC11的治療潛力,發現在腫瘤小鼠模型中,抗PD1單一療法并未提高生存率,而PEGTAT-APC11則延長了生存率。PEG-TAT-APC11和抗PD1的組合進一步改善了生存結果。為了進一步優化腫瘤靶向,作者開發了基于APC11的納米顆粒(NP-APC11)并與腫瘤穿透肽iRGD34綴合以提高選擇性,發現NP-APC11顯著抑制腫瘤生長并增加CD8+T細胞浸潤。作者還評估了NP-APC11與抗PD1聯合治療腫瘤的療效,發現NP-APC11顯著抑制腫瘤生長,NP-APC11與抗PD1聯合治療比抗PD1單一療法具有更有效的治療效果。這些結果證明了APC11單獨或與抗PD1組合作為治療APC缺陷的CRC的有效治療策略的潛力。
圖3 APC11抑制腫瘤生長并增強抗PD1治療的反應
實驗結論
本研究發現PTPN13是APC驅動的免疫逃避的關鍵介質,在機制上APC直接與PTPN13相互作用,阻斷其與STAT1的相互作用,進而抑制腫瘤進展。APC11可以有效阻斷PTPN13-STAT1相互作用,恢復STAT1磷酸化并重新激活針對腫瘤的免疫反應,APC11與抗PD1療法結合時,觀察到更好的腫瘤抑制效果,這為開發CRC患者抗腫瘤藥物提供了新的思路。