
期刊:Journal of Translational Medicine
影響因子:6.1
伯豪技術服務產品:單細胞轉錄組測序
導語
盡管安羅替尼聯合PD-1阻斷療法為晚期非小細胞肺癌(NSCLC)帶來希望,但部分患者仍面臨耐藥難題。最新研究通過單細胞測序分析4.7萬個腫瘤微環境細胞,發現響應患者(MPR)呈現T細胞功能恢復及B細胞- T細胞協同激活,而耐藥患者(Non-MPR)中腫瘤相關巨噬細胞通過VEGF-ZEB1-FLT1軸介導內皮細胞耐藥,關鍵酶PLA2G4A更驅動Treg浸潤形成免疫抑制!該研究為逆轉耐藥提供了精準靶點。
科學問題
1.耐藥機制:盡管聯合療法在臨床試驗中顯示生存獲益,但部分患者仍無響應,其分子和細胞層面的機制尚不明確。
2.腫瘤微環境(TME)動態變化:抗血管生成藥物與免疫治療的協同作用如何通過重塑TME(如免疫細胞、內皮細胞、成纖維細胞的互作)影響療效。
3.VEGF信號通路的作用:VEGF信號如何介導治療響應或耐藥,尤其是與免疫抑制表型(如Treg細胞浸潤)的關聯。
實驗材料:6例晚期NSCLC患者(接受新輔助安羅替尼聯合PD-1阻斷治療后手術切除的腫瘤組織),分為:MPR組(主要病理緩解,3例)、Non-MPR組(非主要病理緩解,3例)
3例治療前(TN)患者的活檢樣本(從基因組序列存檔數據庫下載,編號HRA001033)
主要技術
單細胞轉錄組測序
(技術服務由伯豪生物提供)
研究結果
1. 非小細胞肺癌新輔助治療樣本的單細胞RNA測序分析
本研究收集了6例接受新輔助聯合治療后手術切除的晚期NSCLC患者的腫瘤樣本,通過病理評估分為:MPR組(主要病理緩解,n=3)、Non-MPR組(非主要病理緩解,n=3),另下載3例初治(TN)患者的活檢樣本scRNA-seq數據作為對照。經質控和去重后,最終對47,294個細胞的轉錄組數據進行分析(圖1A)。對所有 9 個病例中的所有細胞進行無監督聚類分析,識別出 23 個簇,根據標記基因的表達,注釋為上皮細胞、內皮細胞、成纖維細胞、T 細胞、B 細胞、平滑肌細胞和髓系細胞(圖 1 B, C)。
為探究治療對腫瘤微環境的重塑作用,作者統計了患者各類細胞比例(圖1D)。在有限病例中發現:Non-MPR患者治療后T細胞比例下降而髓系細胞增加(圖1E)。進一步分析NSCLC中細胞間相互作用,發現上皮細胞通過VEGF信號與內皮細胞互作(圖1F),而髓系細胞與成纖維細胞、內皮細胞及上皮細胞存在強相互作用(圖1G)。
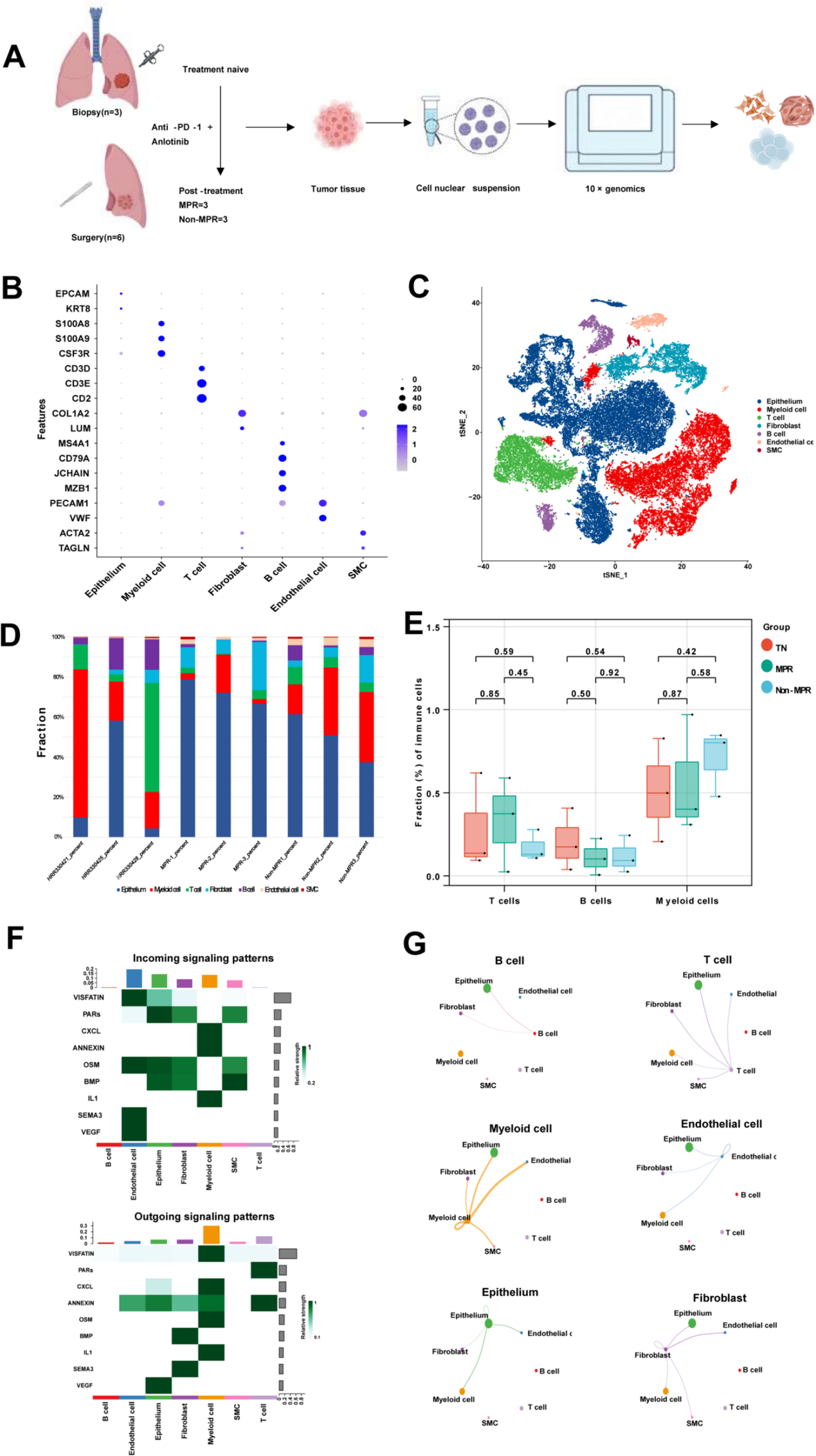
圖1 非小細胞肺癌新輔助治療樣本的單細胞RNA測序分析
2.T細胞向調節性T細胞(Tregs)的轉化提示病理反應不佳
作者重新聚類了 T/NK 細胞,并識別出 7 個簇(圖 2 A, B)。這些簇包括 1 種 T 細胞亞型、1 種 NK 細胞亞型(NK_KLRD1)、1 種 CD8 + T 細胞亞型(CD8_GZMK)、3 種 CD4 + T 細胞亞型(CD4_CCR7、CD4_CD40LG 和 CD4_CD28)以及 1 種 Treg 細胞亞型(Treg_CTLA4)。通過細胞亞群比例比較發現,CD4+ T細胞數量顯著多于CD8+ T細胞。與初治組(TN)和Non-MPR組相比,MPR組患者中高表達IL2RA和TIGIT的Treg_CTLA4細胞減少,而CD4_CCR7細胞增加(圖2C)。進一步鑒定出7個基因(AG3、TIGIT、PCCD1、HAVCR2、CTLA4、LAYN和ENTPD1)構成T細胞耗竭特征標記。治療后,CD4_CCR7細胞的耗竭特征顯著降低(尤其在MPR組)(圖2D),且MPR組的Treg細胞耗竭特征也低于Non-MPR組(圖2D)。代謝特征分析顯示,Treg_CTLA4細胞呈現活躍的丙酮酸代謝,而CD4_CCR7和CD4_CD40LG細胞則以氧化磷酸化為主(圖2E)。多色免疫熒光(mIHC)結果一致表明,Non-MPR組的CTLA4+FoxP3+CD4+ T細胞數量明顯多于MPR組(圖2F),證實治療后MPR患者的Treg細胞比例下降。
同時,T_THEMIS細胞在接受聯合治療后增加(圖2C)。THEMIS是一種參與T細胞激活調控的經典調節蛋白,是T細胞陽性選擇過程中的關鍵因素。與其它細胞簇相比,作者觀察到 T_THEMIS 細胞在氨基酸代謝方面更為活躍,例如“氨基酸-tRNA 生物合成”通路和“纈氨酸、亮氨酸和異亮氨酸降解”通路(圖2E)。因此,作者進行了軌跡分析,以探索T_THEMIS細胞在腫瘤組織中的轉變(圖2G)。從起源(T_THEMIS)到初始T細胞(CD4_CCR7)和活化的調節性T細胞(Treg_CTLA4)存在兩條轉變路徑。分析顯示,T_THEMIS細胞的分化對于Treg細胞的擴增至關重要。作者發現T_THEMIS細胞在偽時間中的分布可以分為兩個狀態。在初級狀態,T_THEMIS細胞高表達DLG2,而在活化狀態下,MHC-II基因(HLA-A和HLA-C)的表達上調。CellPhoneDB分析揭示了整合素介導癌細胞與T_THEMIS細胞之間的通訊(圖2H)。治療后微環境中T_THEMIS細胞轉變的機制需要進一步探索。
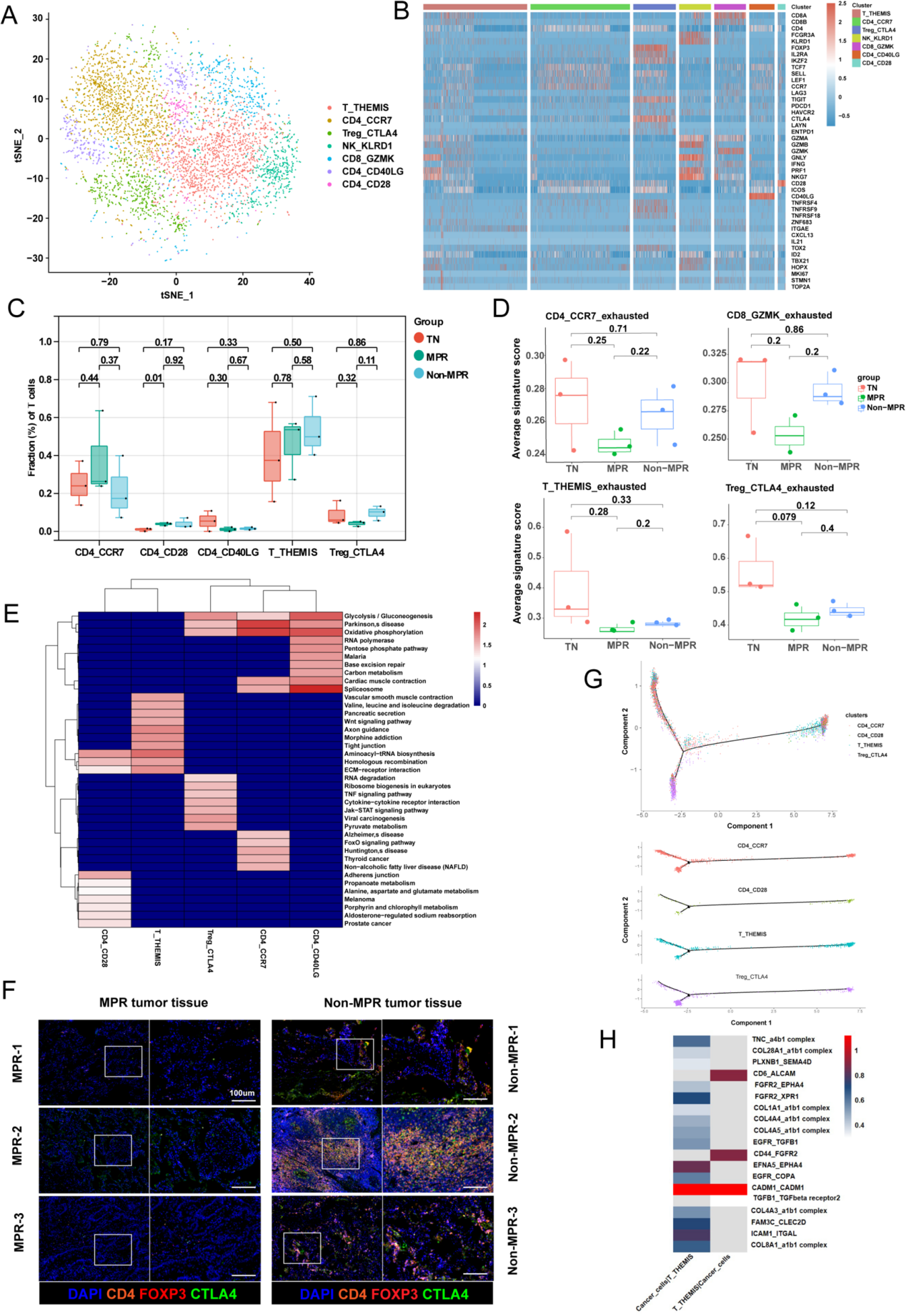
圖2 T細胞向調節性T細胞(Tregs)的轉化提示病理反應不佳
3.PAX5?記憶B細胞與CD4?T細胞之間的正反饋增強了抗腫瘤免疫反應
作者重新對B細胞進行了聚類,將其分為4個簇(圖3A),包括3種漿細胞B細胞亞型(B1_PIM2、B2_JCHAIN和B3_IGHG3)和1種記憶B細胞亞型(B4_PAX5)。隨后,作者比較了各亞型的比例,發現治療后漿細胞B細胞的比例下降,而B4_PAX5記憶B細胞的比例增加,尤其是在主要病理緩解(MPR)患者中(圖3B)。B4_PAX5細胞高表達免疫球蛋白受體(FCRL1)和B細胞功能調節因子(BANK1)。作者發現B4_PAX5細胞富集于“FcγR介導的吞噬作用”通路、“IgA產生”通路和“B細胞受體信號傳導”通路(圖3C)。
此外,mIHC 顯示 CD20 + 細胞在 MPR 患者中比非 MPR 患者更浸潤(圖 3 D)。作者研究了 B4_PAX5 細胞是否可以作為預后的預測指標。在 KMplot 中,作者輸入 PAX5、FCRL1 和 BANK1 進行多基因生存分析,發現較低的 B4_PAX5 特征與不良預后相關(圖 3 E)。這些結果表明 B4_PAX5 細胞的特征可以作為 NSCLC 的預后預測指標。
作者進行了CellPhoneDB分析,以研究T細胞與B4_PAX5細胞之間相互作用的途徑,結果表明B4_PAX5細胞可以通過以下方式與T細胞相互作用:CCL4-CNR2和IFNG-IFNR(圖3F)。這些數據表明,在免疫微環境中,B4_PAX5細胞受到來自CD4_CD40LG細胞的CD40LG和來自CD4_CD28細胞的CD28的驅動,而T細胞則通過來自B4_PAX5細胞的FAM3C被激活,從而增強抗腫瘤免疫反應。
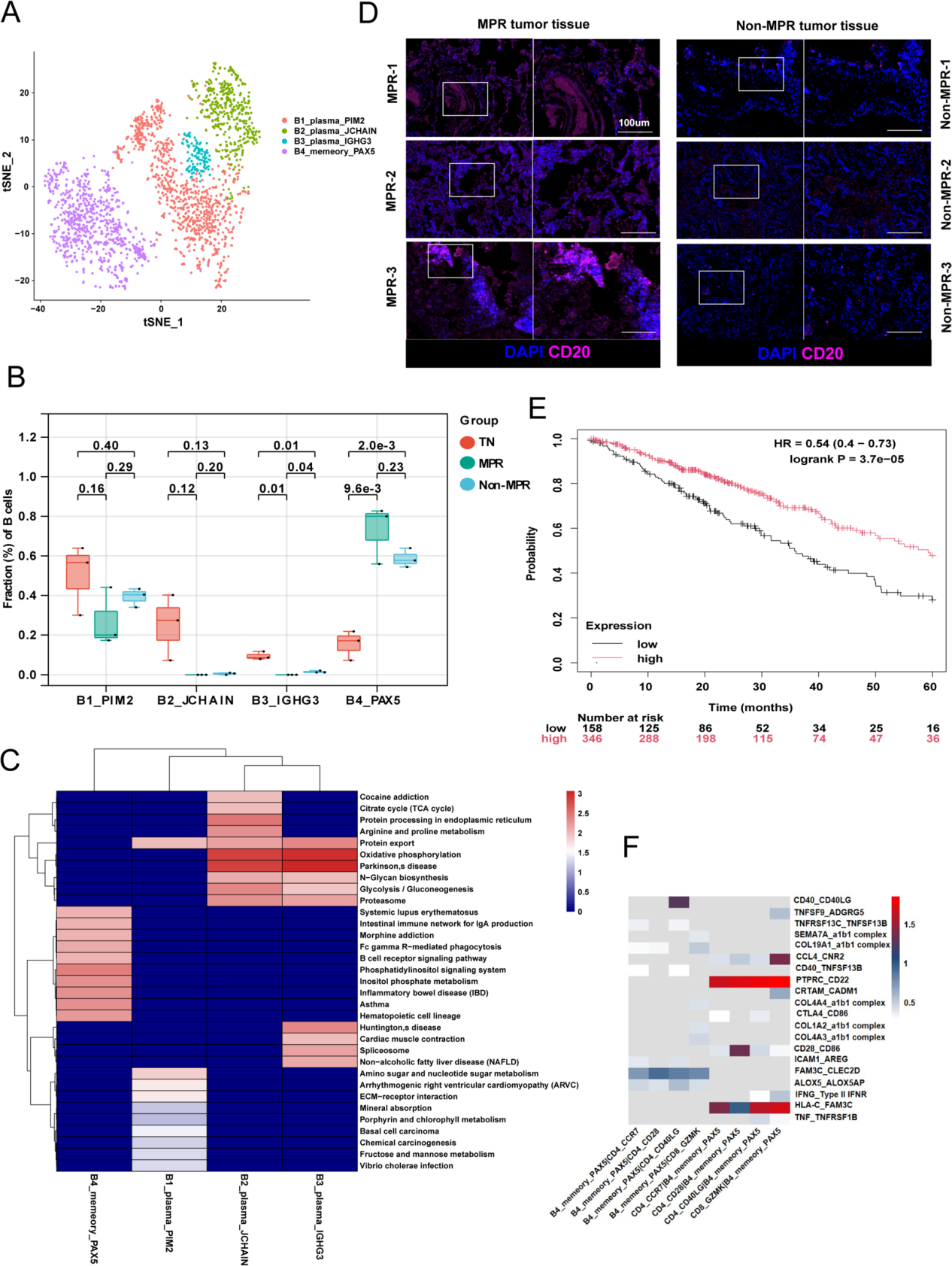
圖3 PAX5?記憶B細胞與CD4?T細胞之間的正反饋增強了抗腫瘤免疫反應
4.腫瘤相關巨噬細胞通過作為細胞通訊樞紐削弱治療效率
髓系細胞組分可劃分為7個亞群,包括2個中性粒細胞亞群、2個巨噬細胞亞群、1個單核細胞亞群、1個樹突狀細胞亞群及1個肥大細胞亞群(圖4A)。其中Mac1_PPARG細胞高表達M2型標志物(MRC1)及經典肺泡巨噬細胞標志物(FABP4和MARCO);而Mac2_CD163L1細胞則高表達HS3ST2與LGMN,并呈現更強的免疫抑制特征(C1QA、C1QB及APOE;圖4B、C)。值得注意的是,治療后Mac1_PPARG與Mac2_CD163L1巨噬細胞均有所增加,且Mac2_CD163L1巨噬細胞在非病理完全緩解(Non-MPR)患者中增幅尤為顯著(圖4D)。進一步通過多重免疫組化(mIHC)證實,CD68+細胞在Non-MPR患者中的浸潤程度高于MPR患者(圖4E)。
中性粒細胞可劃分為2個亞群:1個成熟亞群(CXCR2高表達CXCR4低表達,標記為Neu1_KCNJ15)和1個衰老亞群(CXCR2低表達CXCR4高表達,標記為Neu2_CXCL10;圖4A、B)。其中,成熟亞群Neu1_KCNJ15高表達顆粒蛋白標志物(S100A8和S100A9)及中性粒細胞胞外誘捕網(NETs)標志物(LCP1),這些分子釋放后在炎癥通路中起關鍵作用;而衰老亞群Neu2_CXCL10則過表達多種因子(包括CXCL9、CXCL10和VEGFA)。治療后,成熟Neu1_KCNJ15中性粒細胞數量顯著減少(圖4D)。
單核細胞亞群Mon_VCAN高表達單核細胞標志物(FCN1和VCAN)及更高水平的MHC-II分子(HLA-DRA、HLA-DPA1和CD74),提示其成熟狀態;此外,該亞群還高表達TGFB1,呈現免疫抑制表型(圖4B)。
此外,基于殼層圖譜(shell map),作者對Mac2_CD163L1巨噬細胞與其他細胞間的互作進行分析,以探究治療對巨噬細胞的重塑作用(圖1G)。研究發現,Mac2_CD163L1巨噬細胞分泌的CXCL8、CCL3和IL18,分別通過CXCR1、CCR3和IL18R受體與中性粒細胞發生相互作用。同時,該巨噬細胞亞群可能通過分泌SPP1和CXCL2抑制T_THEMIS細胞及腫瘤細胞,并通過整合素(integrins)與成纖維細胞進行通訊(圖4F)。這些結果表明,Mac2_CD163L1巨噬細胞不僅抑制T_THEMIS細胞和中性粒細胞的功能,還與癌癥相關成纖維細胞存在密切關聯。
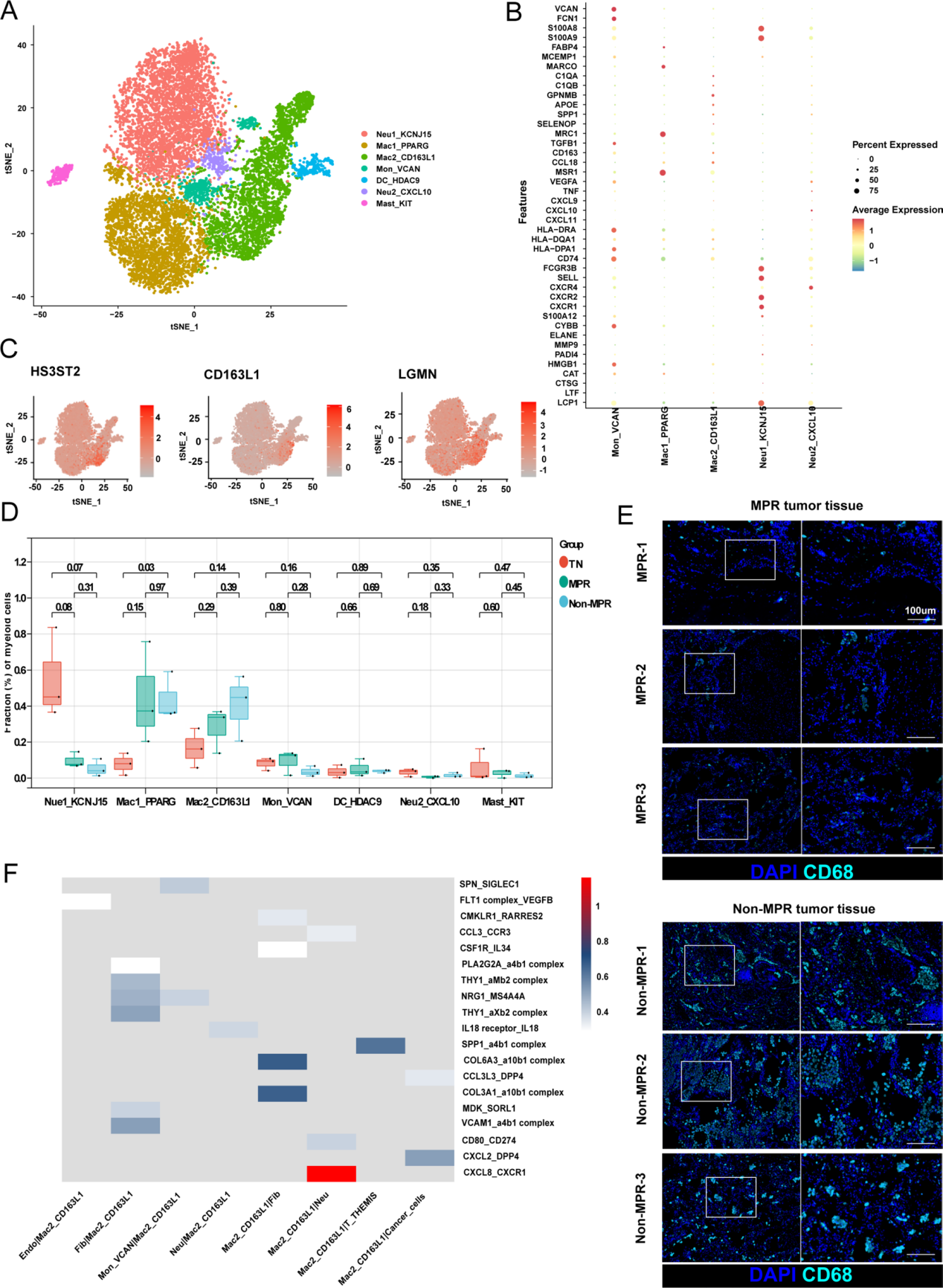
圖4:腫瘤相關巨噬細胞通過作為細胞通訊樞紐削弱治療效率
5. 內皮細胞可能通過ZEB1調控FLT1以響應安羅替尼治療
間質成分可劃分為8個亞群,包括2個內皮細胞亞群、5個成纖維細胞亞群和1個平滑肌細胞亞群(圖5A)。作為安羅替尼的重要作用靶點,作者通過比較內皮細胞亞群比例來探究聯合治療對內皮細胞的重塑效應。
Endo1_FLT1細胞為血液內皮細胞,高表達FLT1(VEGFR)、EGFL7和PECAM1,且在非病理完全緩解(Non-MPR)患者中富集(圖5B、C)。與此同時,Endo2_CD36內皮細胞總占比僅約5%,顯著低于VEGFR陽性(Endo1_FLT1)細胞。這些結果表明,Non-MPR患者內皮細胞中VEGFR的表達與安羅替尼耐藥性密切相關。
GSVA分析顯示,病理完全緩解(MPR)患者的內皮細胞中氨基酸代謝通路顯著富集,而非病理完全緩解(Non-MPR)患者的內皮細胞則呈現原發性免疫缺陷特征(圖5D)。由于內皮細胞是外周免疫細胞與腫瘤細胞間的主要界面,負責傳遞免疫信號并呈遞抗原表位,這一結果表明,Non-MPR患者的內皮細胞可能通過下調抗原呈遞能力和免疫細胞歸巢活性,促進腫瘤免疫逃逸表型,而聯合治療的療效可能依賴于內皮細胞的正常化。
進一步利用SCENIC分析MPR與Non-MPR內皮細胞的差異轉錄因子及其靶基因(圖5E),發現ZEB1在Non-MPR內皮細胞中高表達(圖5F)。ZEB1不僅與免疫微環境中抑制性免疫細胞的增加相關,其結合位點還存在于VEGFR調控基因中,提示ZEB1可能通過調控VEGFR增強內皮細胞的免疫抑制表型。此外,研究發現內皮細胞可被成纖維細胞和Mac2_CD163L1巨噬細胞分泌的VEGF激活(圖4F、5G),表明微環境中存在免疫抑制性細胞通訊反饋環路。在共培養體系中驗證發現,安羅替尼可上調巨噬細胞的VEGFB(而非VEGFA)表達,并促進內皮細胞中VEGFR(FLT1)和ZEB1的表達。
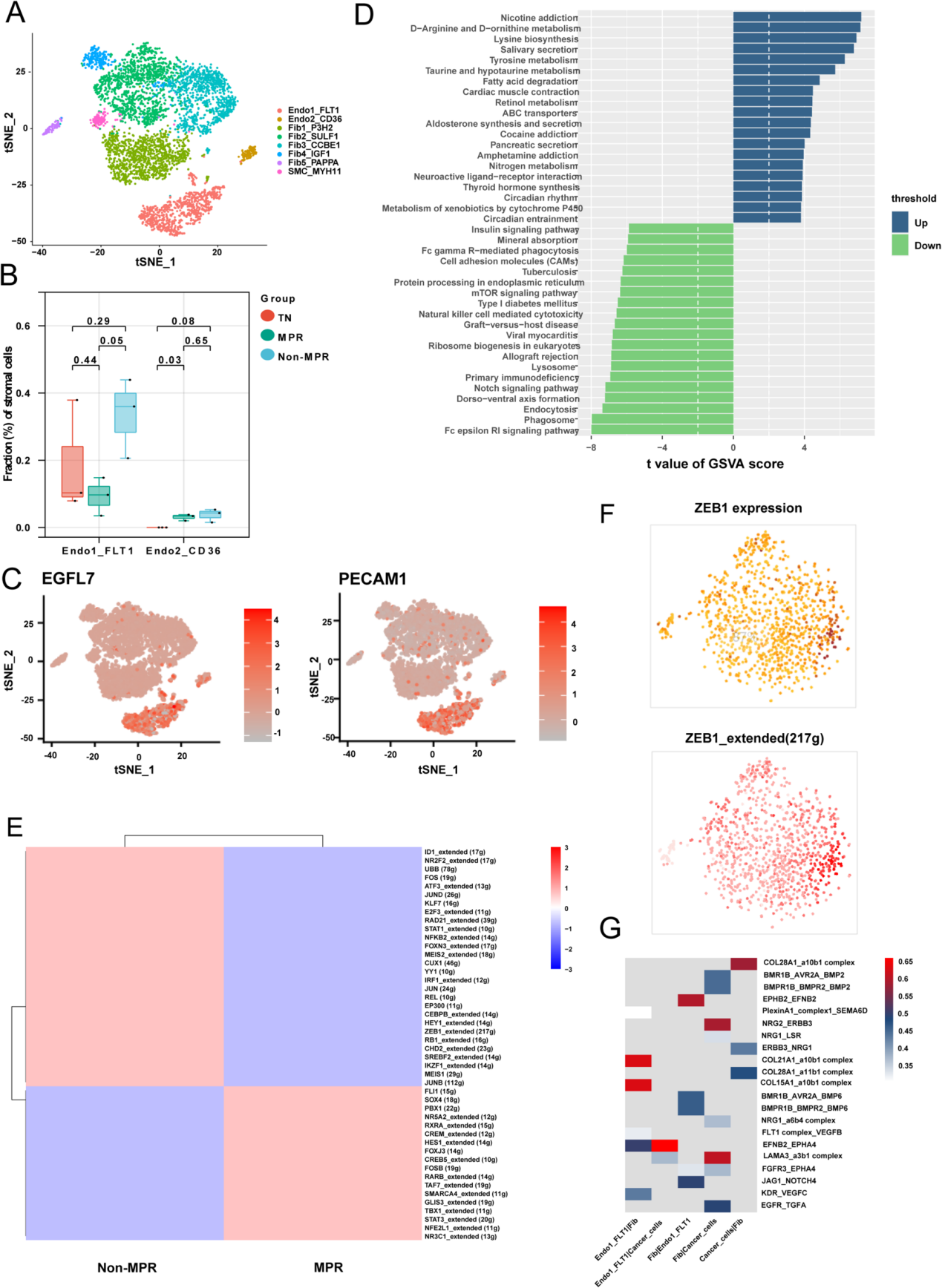
圖5 內皮細胞可能通過ZEB1調控FLT1對安羅替尼的治療響應
6. cPLA2促進調節性T細胞(Tregs)浸潤并誘導耐藥性
作者采用InferCNV分析從7個上皮細胞亞群中區分惡性與正常細胞(圖6A)。除部分E1_ABCA3和E3_TP63細胞外,聚類分析顯示E4_ACSL4與E5_BCAT1亞群被判定為癌細胞。治療后,Non-MPR(非病理完全緩解)患者中E4_ACSL4和E5_BCAT1細胞比例顯著增加。
通過富集分析,作者發現與非腫瘤組織(TN)和病理完全緩解(MPR)患者相比,非病理完全緩解(Non-MPR)患者中VEGF信號通路相關酶(PLA2G4A、PLA2G12A和PTK2)表達顯著升高(圖6B,C)。作者推測,腫瘤細胞可能通過上調PLA2G4A(cPLA2)來應對安羅替尼治療對VEGF通路的抑制作用。
在共培養體系中驗證VEGF信號分子表達變化時發現,與巨噬細胞共培養后,腫瘤細胞中PTK2、PLA2G4A和PLA2G12A表達上調,且安羅替尼處理增強了這一效應。PLA2家族酶能催化膜磷脂水解釋放花生四烯酸,后者可進一步代謝為類二十烷酸物質,參與調控炎癥反應和巨噬細胞極化。
進一步分析cPLA2在免疫微環境中的作用發現,cPLA2表達與FoxP3呈正相關,與CD4呈負相關(圖6D,E)。此外,TCGA數據顯示cPLA2高表達與不良預后相關,作者的NSCLC隊列研究也證實PLA2G4A是一個獨立預后因素(圖6F,G)。這些結果表明,以PLA2G4A為代表的PLA2可能通過促進調節性T細胞(Tregs)浸潤參與免疫抑制,作者推測花生四烯酸代謝產物可能通過腫瘤相關巨噬細胞(TAMs)的SPP1信號通路,促進T_THEMIS細胞向Tregs轉化(圖4F)。
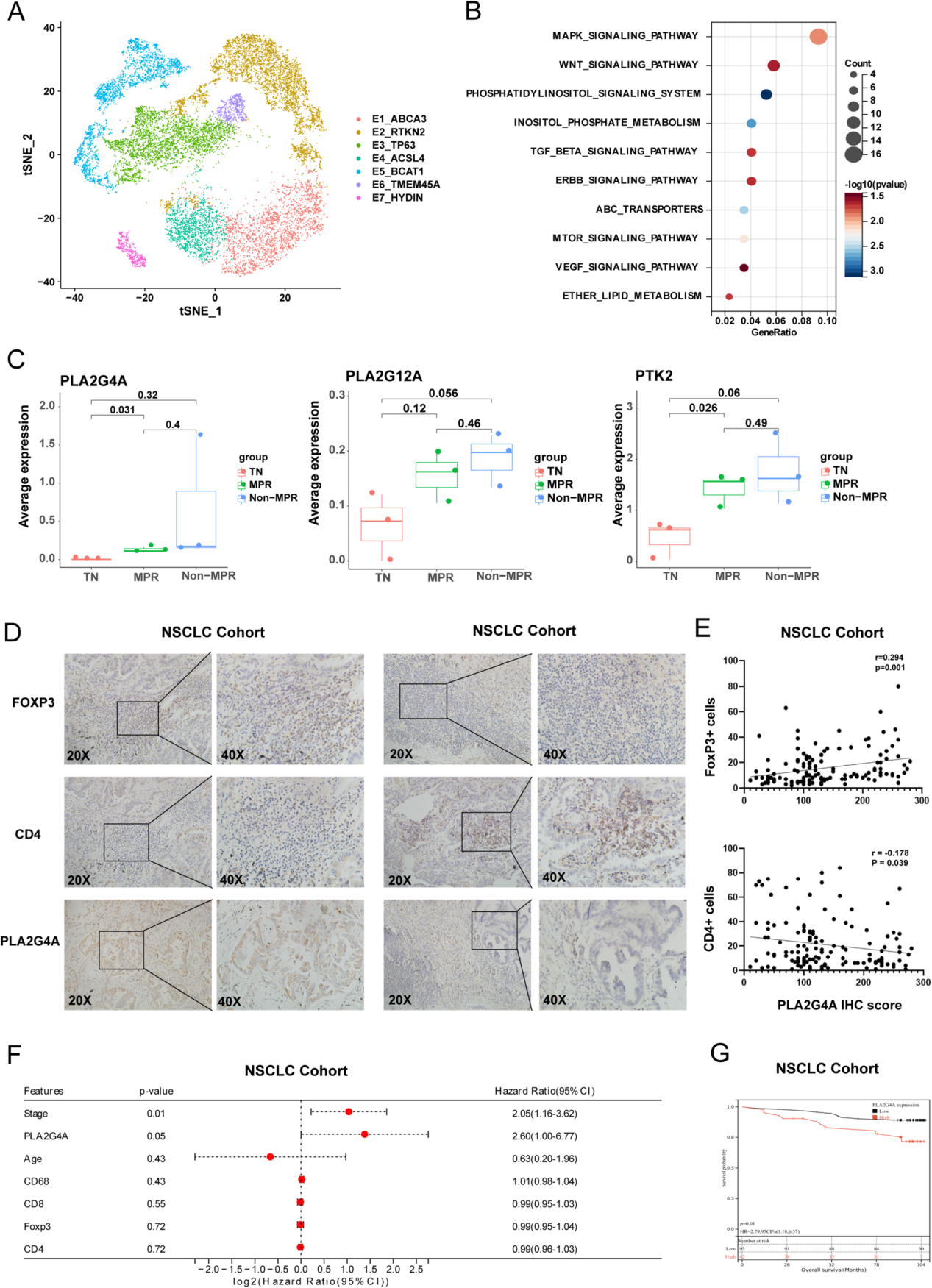
圖6 cPLA2促進調節性T細胞(Tregs)浸潤并誘導耐藥性
研究結論
在這項研究中,作者構建了接受新輔助安羅替尼聯合 PD-1 阻斷治療的非小細胞肺癌患者的單細胞轉錄組圖譜,揭示了腫瘤免疫微環境中的細胞群體異質性和治療失敗的可能潛在機制。作者發現,VEGF 信號通路依賴的血管內皮細胞和上皮細胞的動態變化對非小細胞肺癌患者安羅替尼耐藥性和免疫抑制表型的形成具有深遠影響。
參考文獻:
Huang Z, Li L, Zhao X, Jin H, Shen M, Li B, Zeng Y, Zhang Q, Wang Q, Wang M, Yang L. scRNA-seq reveals that VEGF signaling mediates the response to neoadjuvant anlotinib combined with PD-1 blockade therapy in non-small cell lung cancer. J Transl Med. 2025 Apr 25;23(1):478. doi: 10.1186/s12967-025-06485-4. PMID: 40281576; PMCID: PMC12032801.