編者按
2026年2月28日,第15個國際罕見病日,全球已知罕見病超過7000種,影響約3億人口,其中約80%由遺傳因素導致。僅在中國,罕見病患者總數預計已超過2000萬。然而,由于患者群體分散、病理機制復雜,90%以上的罕見病至今尚無有效治療藥物。2月19日,國際頂級期刊《Nature》發表重磅研究成果,針對罕見病“確診難、漏診率高”的全球性難題,上海交通大學人工智能學院與醫學院附屬新華醫院聯合團隊成功開發了全球首個 AI 智能體罕見病循證推理診斷系統——DeepRare,首次在罕見病診斷的準確性上,超越了擁有十年以上經驗的臨床專家。然而,精準診斷只是第一步,明確致病基因后,如何理解疾病機制?如何篩選驗證潛在的治療藥物?……
近年來,斑馬魚憑借其獨特優勢,在罕見病機制研究、藥物篩選評價、疾病治療等領域展現出巨大的潛力,先后已有數十篇基于斑馬魚模型的罕見病研究成果在nature等頂刊相繼發表。本期,我們一起來回顧2025年斑馬魚在罕見病研究與治療中的應用進展——
01、斑馬魚等揭示靶向ABHD18或可治療巴氏綜合征

文章題目
Genetic suppression features ABHD18 as a Barth syndrome therapeutic target
雜志:Nature(IF=48.5)
發表時間:2025年9月3日
作者:Jason Moffat,Vincent A. Blomen,Sebastian M. B. Nijman等
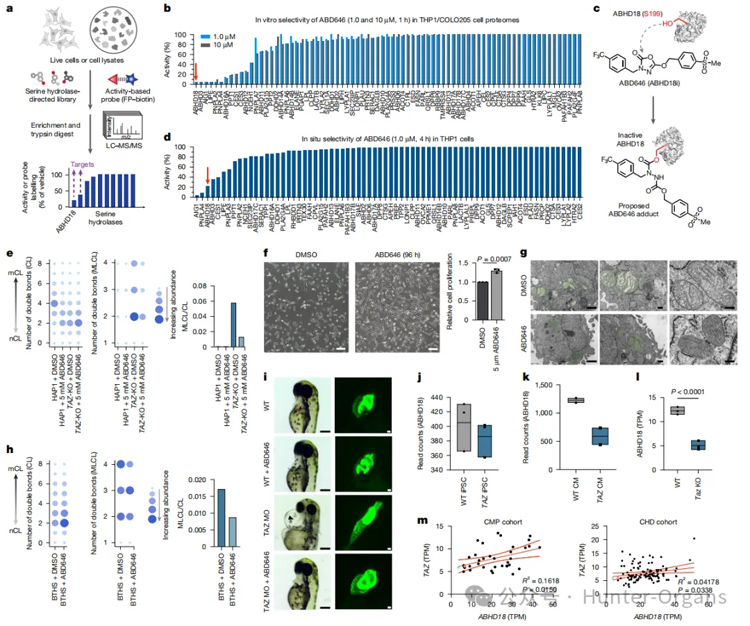
巴氏綜合征(Barth Syndrome, BTHS)是一種罕見的X連鎖遺傳病,主要由TAFAZZIN(TAZ)基因突變引起。TAZ基因負責參與心磷脂(Cardiolipin, CL)的代謝重塑過程。本研究首次確定了ABHD18是人類細胞中催化心磷脂去酰化為MLCL的關鍵酶。研究表明,在TAZ缺陷背景下,通過遺傳敲除或小分子抑制ABHD18,可顯著降低MLCL/CL比率,恢復線粒體超復合物穩定性和能量代謝功能,從而有效緩解小鼠心肌病表型及患者細胞的功能異常。研究團隊進一步開發出了選擇性共價抑制劑ABD646,在患者成纖維細胞、TAZ敲除斑馬魚及Taz−/Y小鼠中均實現疾病表型逆轉,不僅揭示了心磷脂重塑通路的核心節點,還為BTHS的治療提供了全新視角。
02、斑馬魚揭示VACTERL綜合征新機制

文章題目
Loss of med14 causes developmental malformations characteristic of VACTERL association by disrupting the Mediator complex
雜志:Genes & Diseases(IF=9.4)
發表時間:2025年8月2日
作者:Jingwen Liu, Ying Yi Zhang, Qiang Wang等
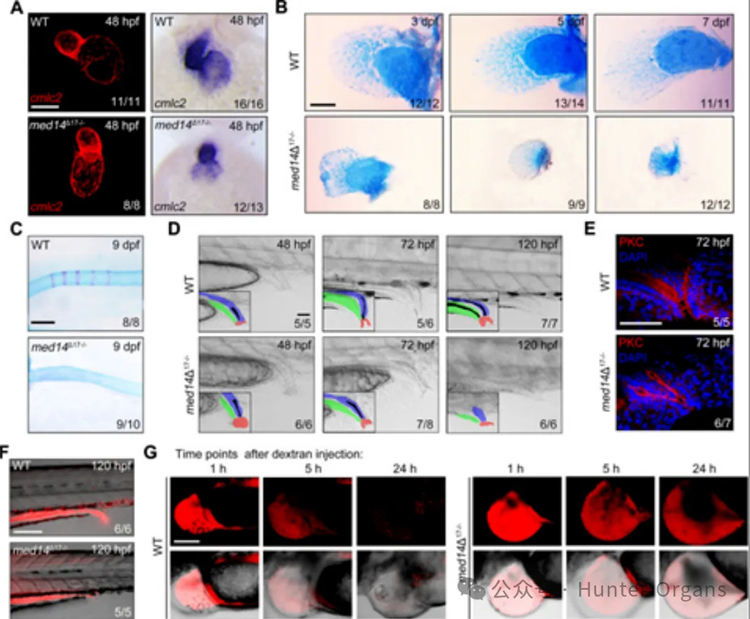
VACTERL綜合征是一種以多器官發育畸形為特征的罕見疾病,包括心臟、胸椎、腎臟等結構異常,但其遺傳病因尚不完全明確。本研究利用斑馬魚模型,揭示了MED14基因在胚胎器官發育中的關鍵作用,并首次發現其突變與VACTERL綜合征的直接關聯。
研究結果顯示,med14基因缺失導致斑馬魚出現心臟、胸鰭、脊椎、泄殖腔和前腎等多器官發育畸形,這些表型與人類VACTERL綜合征的臨床表現高度重合。進一步機制研究表明,Med14是Mediator復合物的核心亞基,其缺失破壞了Mediator復合物的組裝,進而干擾了Wnt信號通路等相關發育信號的轉導。研究團隊在一名VACTERL綜合征患兒及其家族中鑒定出MED14基因的新錯義突變,首次從遺傳學角度確認MED14突變是該罕見病的致病因素之一,為VACTERL綜合征的分子診斷提供了新的候選基因,也為該病的發病機制研究奠定了重要基礎。
03、清道夫內皮細胞通過吞噬溶血有毒分子減輕組織損傷

文章題目
Scavenger endothelial cells alleviate tissue damage by engulfing toxic molecules derived from hemolysis
雜志:PNAS(IF=9.4)
發表時間:2025年2月11日
作者:Yimei Dai,Jin Xu等
文章主題:
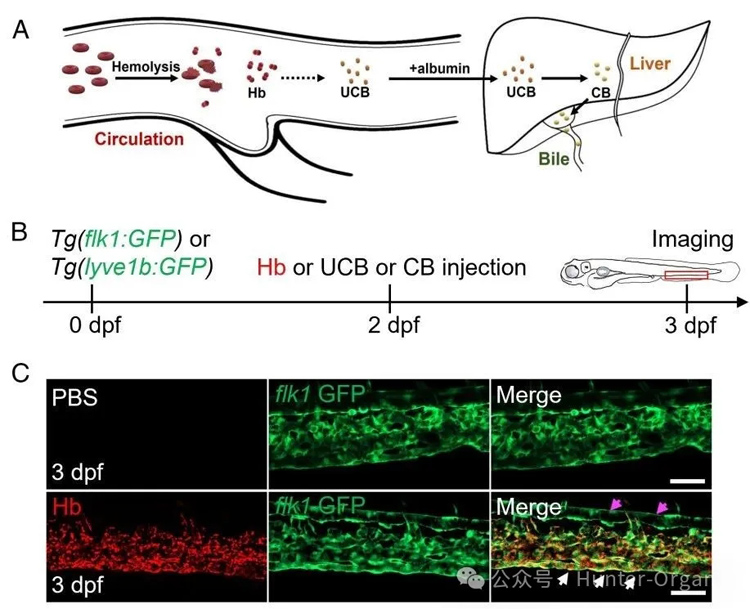
紅細胞生成性卟啉病(EPP)是一種光誘導溶血的罕見遺傳病,主要癥狀是皮膚光敏性和肝臟疾病。本研究在斑馬魚中發現清道夫內皮細胞(SECs),一種具有顯著內吞能力的特化內皮細胞,能夠吞噬溶血的兩種主要有毒副產物——大分子血紅蛋白(Hb)和小分子未結合膽紅素(UCB),其在生理及病理性溶血過程中起保護性作用。為了證明SECs在溶血過程中的保護作用,研究團隊利用斑馬魚紅血球生成性卟啉癥模型,該模型由于鐵螯合酶突變導致過量的原卟啉IX(PPIX)積累,從而引起光敏感性溶血和幼蟲死亡。SECs通過Stab2促進過量PPIX的清除,從而減輕了PPIX引起的幼蟲死亡。
04、利用斑馬魚Werner綜合征模型發現潛在抗衰藥

文章題目
Establishment and application of a zebrafish model of Werner syndrome identifies sapanisertib as a potential antiaging drug
雜志:PNAS(IF=9.4)
發表時間:2025年1月30日
作者:Jianlong Ma,Jingmei Song,Lingfei Luo 等
文章主題:
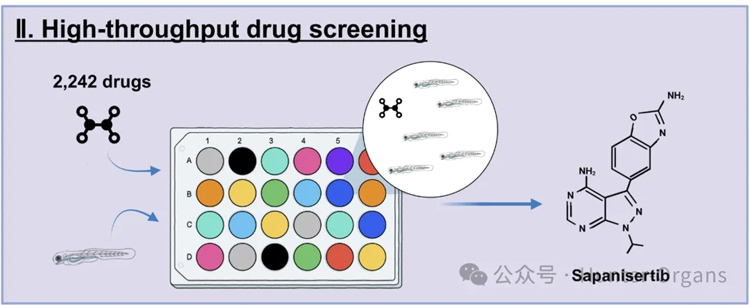
Werner綜合征是一種罕見的遺傳性早衰疾病,由WRN基因突變引起,常表現出基因組不穩定、細胞衰老和壽命縮短等衰老特征。本研究通過建立斑馬魚Werner綜合征模型,成功構建了一種高效、經濟的斑馬魚抗衰老藥物篩選平臺,并從小規模篩選中發現抗癌藥物sapanisertib具有延緩衰老的潛力,不僅為早衰癥患者提供了潛在的治療方案,也為延緩衰老提供了全新的藥物和機制。
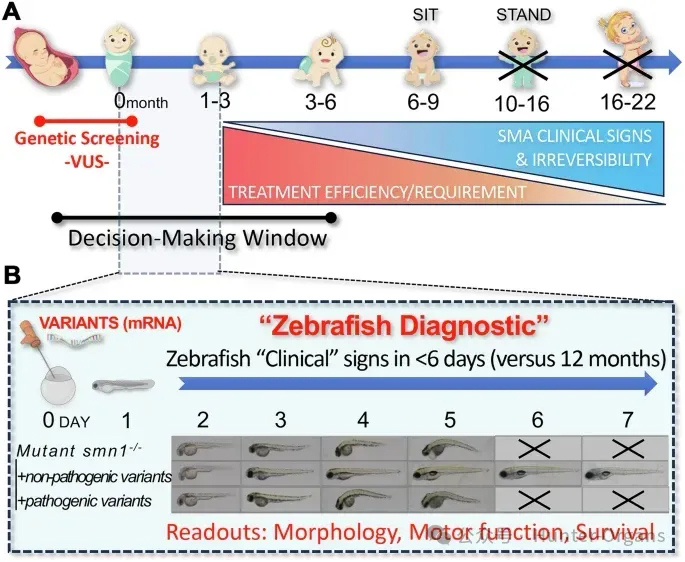
05、基于SMN1/SMA驗證探究斑馬魚基因變異的臨床相關性

文章題目
Clinical relevance of zebrafish for gene variants testing. Proof-of-principle with SMN1/SMA
雜志:EMBO Molecular Medicine(IF=8.3)
發表時間:2025年12月15日
作者:Brett W Stringer, Brunhilde Wirth, Jean Giacomotto等
文章主題:
脊髓性肌萎縮癥(SMA)是一種由SMN1基因功能喪失導致的遺傳性疾病,表現為脊髓和腦干運動神經元丟失,進而引起進行性肌無力和萎縮。SMA曾是全球嬰兒死亡最常見的遺傳原因。本研究首次證明了斑馬魚可作為臨床基因變異解讀的高效工具。針對新生兒篩查中發現的兩種SMN1意義不明確變異(VUS),研究團隊利用斑馬魚SMA模型在18個工作日內完成了功能性評估。
研究結果顯示,兩種VUS mRNA均可完全挽救smn基因敲除斑馬魚的形態缺陷、運動功能障礙和早期死亡表型,與野生型SMN1 mRNA效果相當。基于這一結果,臨床團隊決定對兩名攜帶VUS的嬰兒暫不啟動治療。截至14月齡,兩名兒童仍無癥狀,生長發育正常,避免了每人超過200萬美元的不必要治療費用。研究還證實該方法可區分部分功能喪失的低效等位基因,為晚發型SMA的診斷提供參考,也為斑馬魚模型在臨床基因變異解讀中的應用提供了堅實的原理驗證,表明其可作為精準醫療時代支持臨床決策的有力工具。
作為健康美麗產業CRO服務開拓者與引領者、斑馬魚生物技術的全球領導者,環特生物搭建了“斑馬魚、類器官、哺乳動物、人體”四位一體的綜合技術服務體系,開展健康美麗CRO服務、科研服務、智慧實驗室搭建三大業務。目前,環特已建立Werner綜合征、先天性脊柱側凸、Acrofacial Dysostosis–Cincinnati 綜合征(辛辛那提型肢面發育不良,AFDCIN)、Dravet綜合征(DS)等斑馬魚罕見病模型及200多種各類疾病斑馬魚模型,腦類器官、心臟類器官及各種腫瘤類器官培養平臺,歡迎有需要的讀者垂詢!